


The advent of Milasen, a groundbreaking patient-customized antisense oligonucleotide therapy, has heralded a new era in personalized medicine for rare genetic disorders. This pioneering approach, exemplified by Milasen and its successors, not only offers hope to patients with previously untreatable conditions but also raises critical questions about the regulatory framework governing such individualized treatments. As the field of patient-specific oligonucleotide therapies rapidly evolves, it becomes increasingly crucial to comprehend the FDA's guidances and their far-reaching implications for future N-of-1 clinical trials. TriApex, leveraging its extensive experience in nonclinical research on oligonucleotide drugs, has proactively initiated nonclinical toxicology studies for certain projects focusing on Individualized Antisense Oligonucleotide Drug Products for Severely Debilitating or Life-Threatening Diseases (SDLT). These studies, conducted in accordance with FDA guidelines, represent a significant effort to expedite the commencement of clinical trials for these innovative therapies. This comprehensive review begins with Mila's compelling story and further explores the regulatory considerations emerging from FDA guidances related to patient-customized oligonucleotide therapies. We provide an in-depth analysis of key aspects outlined in these guidances, including the intricate design of N-of-1 clinical trials, essential nonclinical studies, and critical Chemistry, Manufacturing, and Controls requirements. By dissecting the regulatory landscape, we aim to elucidate a clear path forward for making these innovative, tailored therapies more accessible to patients with rare genetic conditions. By bridging the gap between cutting-edge science and regulatory frameworks, we strive to accelerate the development and delivery of life-changing treatments to those who need them most, potentially transforming the landscape of rare disease management.
Mila's Story: A Race Against Time Timeline from the initial clinical diagnosis to the initiation of treatment[1]
The Disease Discovery In 2016, a 6-year-old girl named Mila began showing signs of a rare and fatal neurodegenerative condition. Her symptoms started subtly at age 3 with an inward-turning foot, progressed to vision problems at 4, and by 5, she was experiencing language regression and clumsiness. As she approached her 6th birthday, Mila's condition rapidly deteriorated, leading to hospitalization due to vision loss, frequent falls, and difficulty speaking and swallowing. After extensive testing, a skin biopsy revealed abnormal lysosomal inclusions characteristic of Batten's disease. Genetic testing identified one known pathogenic mutation in the MFSD8 gene, but the second mutation remained elusive. Determined to find answers, Mila's family enrolled her in a research study at Boston Children's Hospital.
The Breakthrough Using whole-genome sequencing (WGS), researchers discovered a novel mutation: an SVA retrotransposon insertion in MFSD8 intron 6. This insertion caused missplicing of the gene, leading to Mila's condition. With the genetic cause identified, the race was on to develop a treatment.
The Birth of Milasen Inspired by the success of nusinersen, an antisense oligonucleotide (ASO) for spinal muscular atrophy, researchers designed a custom ASO to correct Mila's specific mutation. After testing multiple candidates, they identified the most effective one, naming it "milasen" after Mila. Milasen Development[1]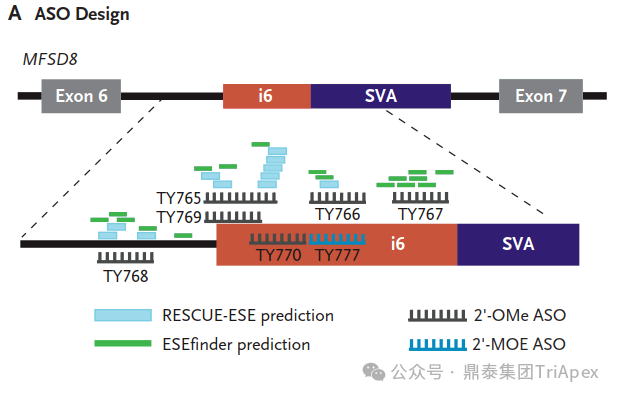
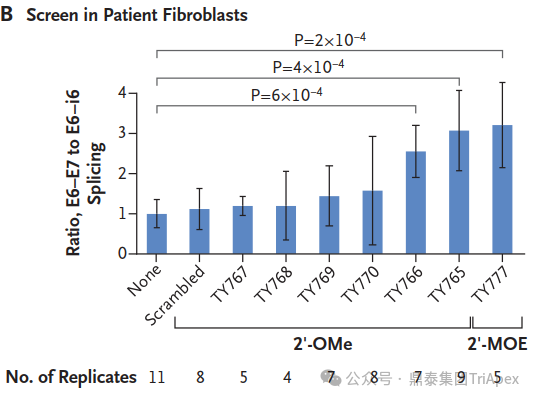
From Lab to Treatment in Record Time In an unprecedented feat, researchers moved from genetic discovery to clinical treatment in just one year. They conducted rapid toxicology studies, manufactured the drug, and obtained FDA approval for an expanded access investigational new drug application.
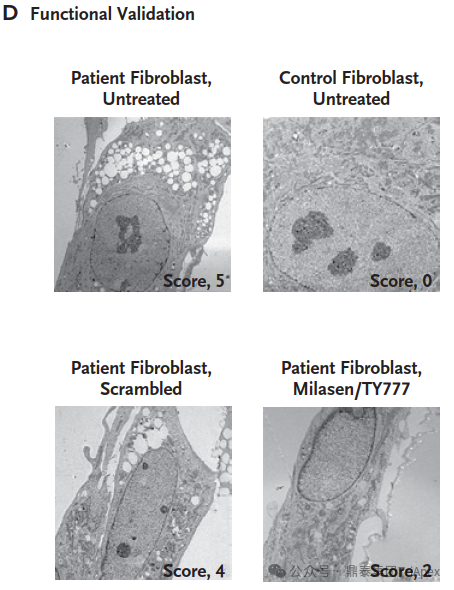
A Glimmer of Hope Mila began receiving milasen treatments in January 2018, when she was 7 years old. The drug was administered intrathecally, with doses gradually increasing over time. Within the first year of treatment, Mila showed no SAEs, and her seizure frequency and duration decreased significantly. The dosing schedule of milasen[1]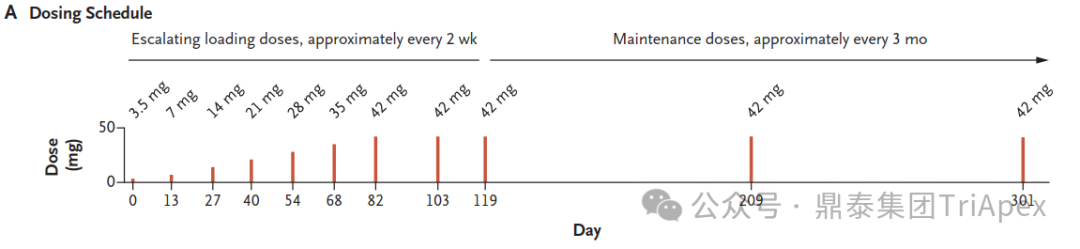
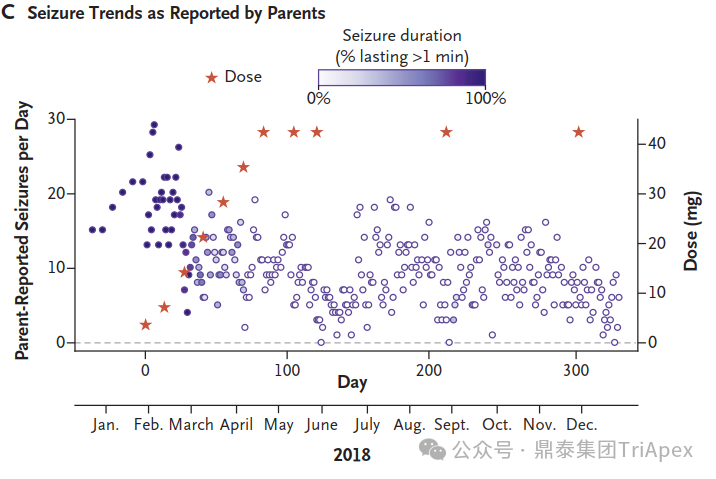
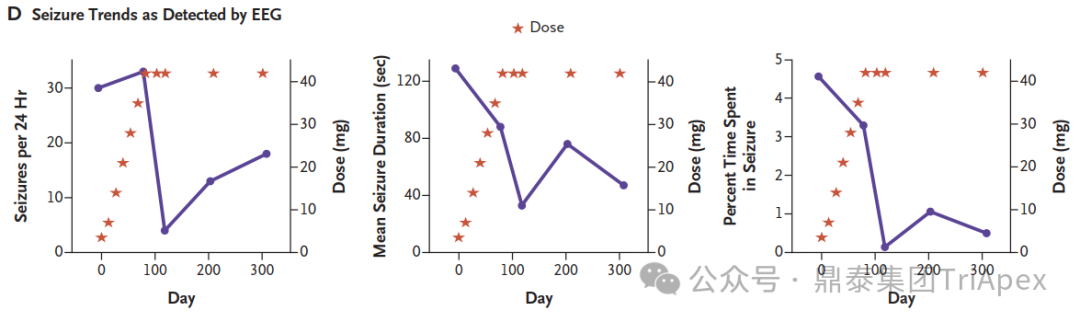
The concentration of milasen in cerebrospinal fluid (CSF) before each administration (trough) and at day 174 without concurrent dose administration. [1] The trends in seizure frequency and duration as reported in a seizure diary recorded by the parents[1] The trends in seizure activity as detected by electroencephalography (EEG)[1] While Mila's story represents a groundbreaking approach to personalized medicine for rare genetic diseases, it also highlights the challenges and ethical considerations in developing patient-specific treatments. Mila's journey offers hope for other patients with rare genetic conditions and paves the way for future advancements in tailored therapies.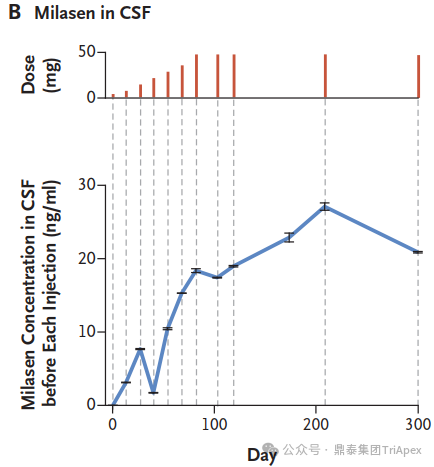
Part I:IND Submissions for Individualized Antisense Oligonucleotide Drug Products for Severely Debilitating or Life-Threatening Diseases: Clinical Recommendations Guidance for Sponsor-Investigators The guidance provides recommendations for managing administration, dosing, and clinical monitoring of individualized ASO drug products for SDLT genetic diseases with no alternative treatments. It focuses on ASOs targeting unique genetic variants specific to 1-2 patients, not larger patient populations with same disease[2]. 1 Ethical and Human Subject Protection ▶The protocol must be reviewed and approved by an institutional review board (IRB) before proceeding, per 21 CFR part 56. ▶Informed consent must be obtained, including risks, potential benefits, and providing updates. ▶If the individualized ASO drug product is intended for use in children, there are some special considerations: The requirements under 21 CFR part 50, subpart D (Additional Safeguards for Children in Clinical Investigations) must be met. This subpart provides additional protections for children involved as subjects in clinical investigations, including obtaining appropriate informed consent/assent, and having procedures to respect child dissent when applicable. The IRB review is critical for studies involving children. 2 Diagnostic and Genetic Considerations ▶Confirm the patient's clinical and genetic diagnosis through relevant laboratory testing(e.g., gene sequencing, enzymatic analysis, biochemical testing, imaging evaluations, as applicable). ▶Provide evidence linking the targeted gene variant(s) to the patient's disease pathogenesis. If the evidence is insufficient, additional investigation(s) may be needed before initiating use of the ASO drug product. ▶Show the targeted variant(s) are unique to the patient, including: prevalence estimates of the targeted gene variant(s) in the disease the projected incidence of the targeted variant(s) in new cases of the disease based on molecular genetic considerations (e.g., type of mutation (missense, frameshift, other), location in the protein). 3 Dosing Considerations ▶ Starting dose Starting dose should be based on available nonclinical data and be scientifically supported by available information from other ASO drug products in the chemical class and/or prior clinical experience. In general, the starting dose should be expected to have a pharmacologic effect, based on available nonclinical in vitro and/or in vivo data. ▶ Dose escalation approach Given the limited nonclinical data available when initiating dosing, the sponsor should consider a dose escalation approach. Dosing should be initiated at a low level and escalated to higher doses cautiously based on: Pharmacodynamic (PD) effects observed Trial participant's response (tolerability or perceived benefit) Available nonclinical data The protocol should clearly outline: The starting dose and justification for selecting it The dosing interval. The plan for dose escalation. The plan for dose reduction in case of toxicity. If a loading dose is proposed, the protocol should contain a rationale for using a loading dose(s) and maintenance dose interval(s), providing a rationale based on: The ASO's half-life. Likely distribution to target areas (e.g. CNS, kidney). Anticipated toxicities at different dose levels. The dose escalation should be done cautiously(e.g.,by no more than a two-fold increment at a time), allowing sufficient time to observe the response to a dose change before making further dose modifications. If nonclinical toxicology studies show a steep dose- or exposure-response relationship for severe toxicities, or if no predictive marker of severe toxicity is available in humans, the sponsor should escalate the dose using smaller increments rather than doubling the dose (e.g., fractional increments rather than dose doubling). In general, concurrent changes to both the dose amount and the dosing interval should be avoided unless there is a justification provided. ▶ Dose de-escalation or discontinuation Dose de-escalation or discontinuation may be necessary to manage unacceptable toxicities and maintain an appropriate benefit-risk profile for the individualized ASO drug product. The protocol should clearly specify a plan for dose de-escalation or discontinuation if a dose-limiting toxicity develops during the trial. Having a pre-specified plan in the protocol allows for proactive management of dose-limiting toxicities that may emerge during the trial. If a decision is made to de-escalate or discontinue the dose due to a dose-limiting toxicity, the sponsor should inform the trial participant or the participant's legally authorized representative about the reason(s) for making such dose changes. 4 Drug Product Administration Procedures ♦ Administer starting dose and escalations in inpatient (hospital) setting with close monitoring initially. ♦ For intrathecal, use experienced personnel and have sedation/interventions available. ♦ For at least 24 hours after administration, the participant should be closely monitored for acute, serious adverse events (AEs) that may be precipitated by: The administration procedure itself (e.g. intrathecal administration) The ASO drug product (e.g. hemodynamic instability, severe hypersensitivity/anaphylaxis, CNS AEs) ♦ After the safety and tolerability at a given dose level is adequately characterized and any AEs are identified and can be appropriately monitored, subsequent doses at the same level can generally be administered in an outpatient (clinic) setting, if needed. Appropriate safety monitoring and interventions should be available in the outpatient setting for possible serious AEs like anaphylaxis. ♦ For intrathecal ASO drug products, experienced medical personnel should perform the intrathecal administration and administer sedation and/or analgesics if needed for the procedure. Appropriate medical monitoring, procedural interventions, and therapeutic interventions should be available in case of complications from the intrathecal procedure and/or sedation. The guidance recommends an inpatient setting for initial doses and dose escalations to closely monitor for serious AEs, with a potential shift to outpatient setting once the safety at a dose level is characterized. It also outlines specific considerations for intrathecal administration. 5 Safety Assessment According to the FDA guidance, the key recommendations for safety assessments of investigational ASO drug products are: ♦ Perform routine safety assessments (e.g. AEs, vital signs, ECG, lab tests) while the ASO is being administered and for a period after stopping, based on the anticipated half-life. ♦ Safety monitoring should be informed by: Nonclinical toxicology findings. Potential off-target gene effects predicted by bioinformatics tools. Previously identified risks with other ASOs of the same class. ♦ Include a detailed schedule of safety assessments in the protocol. ♦ Conduct safety assessments more frequently during the initial dosing period (e.g. first 2-3 months). At minimum, assess before each dose or more frequently if long dosing intervals. ♦ Based on initial results, the frequency of assessments can then be decreased. Clearly outline this in the protocol. ♦ For phosphorothioate (PS) or mixed-backbone ASOs, perform platelet counts at least every 2 weeks or before each dose, whichever is more frequent, due to thrombocytopenia risk. Closely monitor for abnormal findings/bleeding. ♦ For intrathecally administered ASOs, collect cerebrospinal fluid before each dose to evaluate for inflammation/infection. Use remaining samples for PK or biochemical assessments if feasible. ♦ If ASO administration is discontinued for safety reasons, continue safety monitoring until adverse event resolution and evaluate for potential late-onset toxicities. 6 Clinical Response Assessment Assessing whether the ASO drug product is providing clinical benefit to the participant is an important element throughout the administration period. The guideline emphasizes careful tracking of clinical status, use of relevant assessments/biomarkers, continual benefit-risk evaluation, and having criteria for dosing adjustments or discontinuation based on clinical response. To inform decisions on continuing administration, sponsors should carefully track clinical changes over time and have a specific plan for adjusting the dosing regimen to achieve a desirable response. The protocol should clearly outline the specific methods and timing for clinical assessments. Clinical assessments can include outcome scales, performance tests, or patient/caregiver reported symptom changes to follow important disease symptoms during ASO administration. Assessments will depend on the participant's pre-treatment signs/symptoms, underlying disease stage/severity at initiation. Administer assessments at specified intervals determined by disease pathophysiology as well as PK/PD of the ASO drug product. Pharmacodynamic biomarkers (lab tests, imaging, etc.) scientifically justified and relevant to the disease may help assess response, with consideration of assay validation issues. Have a pre-specified plan to continually assess clinical response to ensure benefit-risk remains favorable for continued administration. Consider discontinuing if there is inadequate clinical response or if toxicities outweigh observed clinical benefits. In summary, the key points cover ethical considerations, diagnostic confirmation, dosing strategy, administration procedures, safety monitoring, and assessing clinical response for these individualized ASO drug products in this guidance. Based on this guidance, valuable experience will be accumulated through practice, providing reference for the development of individualized ASO drug products in the future, and further improving this guidance as well. 下期预告:N-of-1 Clinical Trials(Series II)—— Nonclinical Studies 往期精选

Reference: [1] J. Kim, C. Hu, C. Moufawad El Achkar, et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease[J]. N Engl J Med 2019;381:1644-52. [2] FDA. IND Submissions for Individualized Antisense Oligonucleotide Drug Products for Severely Debilitating or Life-Threatening Diseases:Clinical Recommendations. Dec, 2021. [3] FDA. Nonclinical Testing of Individualized Antisense Oligonucleotide Drug Products for Severely Debilitating or Life Threatening Diseases Guidance for Sponsor-Investigators. Apr, 2021 [4] FDA. Investigational New Drug Application Submissions for Individualized Antisense Oligonucleotide Drug Products for Severely Debilitating or Life-Threatening Diseases: Chemistry, Manufacturing, and Controls Recommendations Guidance for Sponsor-Investigators. Dec, 2021







 法规政策
法规政策 项目管理
项目管理


 Global
Global



 苏公网安备32011202001081
苏公网安备32011202001081