


在前几期文章中,鼎泰团队先后对已上市的前 4 款 siRNA 药物进行了较为全面的复盘和分析,分别为 Patisiran(Onpattro®,点击查看原文)、Givosiran(Givlaari®,点击查看原文)、Lumasiran(Oxlumo®,点击查看原文)及 Inclisiran(Leqvio®,点击查看原文)。通过这些内容,我们深入探讨了这 4 款 siRNA 药物的临床开发路径、上市历程和非临床支持临床转化的关键环节,为 siRNA 药物临床研究的设计、结果解读、监管互动的时间节点规划,以及药政策略制定提供了重要的参考和启发。
作为一种有潜力的治疗策略,siRNA 药物受到了业界的广泛关注。鼎泰团队本次分享的是第 5 款获批上市的 siRNA 药物 Vutrisiran,同时也是由 Alnylam 研发上市的第二款针对转甲状腺素运载蛋白淀粉样变性(hATTR-PN)的药物。Vutrisiran 于 2022 年 6 月由 FDA 首次批准上市,此时距第 1 款治疗 hATTR-PN 的药物 Patisiran 上市(2018 年 8 月由 FDA 批准上市)不到 4 年,足以表明 Alnylam 在 hATTR-PN 领域很早就展开了深远的布局和迭代开发。
海日生残夜,江春入旧年。Vutrisiran 在临床开发中展现出显著的潜力,仅通过 1 项 I 期临床研究和 1 项 III 期临床研究(代号 HELIOS-A,中文赫利俄斯,古希腊神话中真正驾着太阳车的太阳神)即支持其上市。这一过程充分体现了其对产品迭代的深刻理解,对前期包括 Patisiran 在内的临床数据和药政策略的熟练运用,以及临床路径的合理优化。
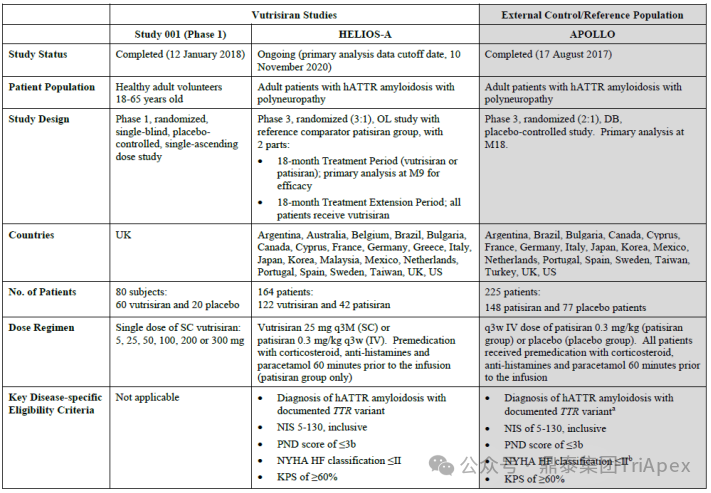
支持 Vutrisiran 上市的两项临床研究
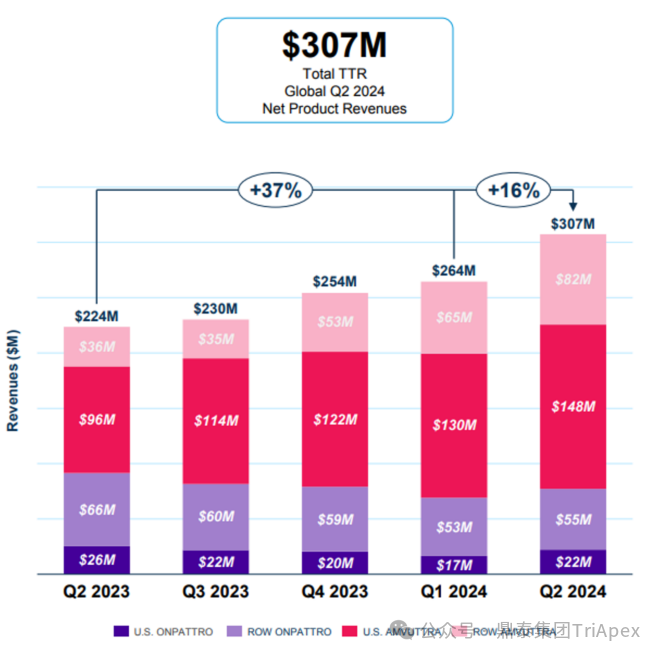
Vutrisiran 上市以来的销售表现 自 Vutrisiran 上市以来,销售势头强劲。根据 Alnylam 公司 2024 年 8 月 1 日披露的半年报:Amvuttra 2024 H1 销售额 4.25 亿美元,同比增长 81.8%;2024 Q2 销售额 2.3 亿美元,同比增长 74%。与此同时,Alnylam 的第一代 hATTR 治疗药物 Patisiran 在 2023 年收入约 3.54 亿美元,同比下降 36%。这一变化主要就是由于患者转向了 Vutrisiran,进一步印证了迭代产品 Vutrisiran 所具有的临床优势。
在本期文章中,我们将进一步探讨 Vutrisiran 的临床开发路径,深入分析其从非临床研究到临床应用的转化过程,并总结其产品迭代的研发思路。作为在寡核苷酸药物研发领域深度赋能的 CRO,鼎泰团队将通过本期内容详细探讨其成功的关键因素,为未来类似药物的非临床研发、临床转化及上市提供宝贵的见解。此外,为了更好地支持当前研发火热的 siRNA 药物更好地进入首次人体试验(FIH),专门总结了已上市 siRNA 药物临床起始剂量设计的基本思路,以期为众多即将进入临床试验的 siRNA 产品的临床研发提供有益的借鉴。
Patisiran 及 Vutrisiran 产品外包装
全文共:11925 字 36 图
预计阅读时长:30 分钟
★ 文章导览 ★
1 | Vutrisiran 研发背景 关键词:转甲状腺素运载蛋白淀粉样变性 |
2 | Vutrisiran 临床开发路径 关键词:临床起始剂量、2项临床试验 |
3 | 监管互动里程碑 关键词:EOP2、Pre-NDA |
4 | 免疫原性检测 |
5 | 非临床与临床试验相关性分析 关键词:非临床研究毒性发现与临床研究相关性、已上市 siRNA 药物临床起始剂量设计 |
6 | 小结 |
Vutrisiran 研发背景
1.1 疾病背景

转甲状腺素蛋白淀粉样变性多发性神经病(transthyretin amyloid polyneuropathy,ATTR-PN)又称转甲状腺素蛋白相关家族性淀粉样变性多发性神经病(transthyretin familial amyloid polyneuropathy,TTR-FAP),是由编码转甲状腺素蛋白的TTR基因致病变异导致的一种罕见的常染色体显性遗传性、以周围神经损害为主的多系统疾病。TTR 蛋白主要在肝脏中产生,通常是维生素 A 的载体。关于该疾病的病因学和流行病学详见 Patisiran(点击查看链接)
1.2 siRNA-TTR 产品开发背景和迭代开发策略
Alnylam 的管线布局主要分为 5 大类:基因遗传病、心血管代谢疾病、传染病、中枢神经系统(CNS)、眼科疾病。ATTR-PN 是 Alnylam 最早开发成药的疾病,也是目前 Alnylam 上市产品最多、产品开发效率较高的方向,可以说是 Alnylam 探索新技术的“试金石”。
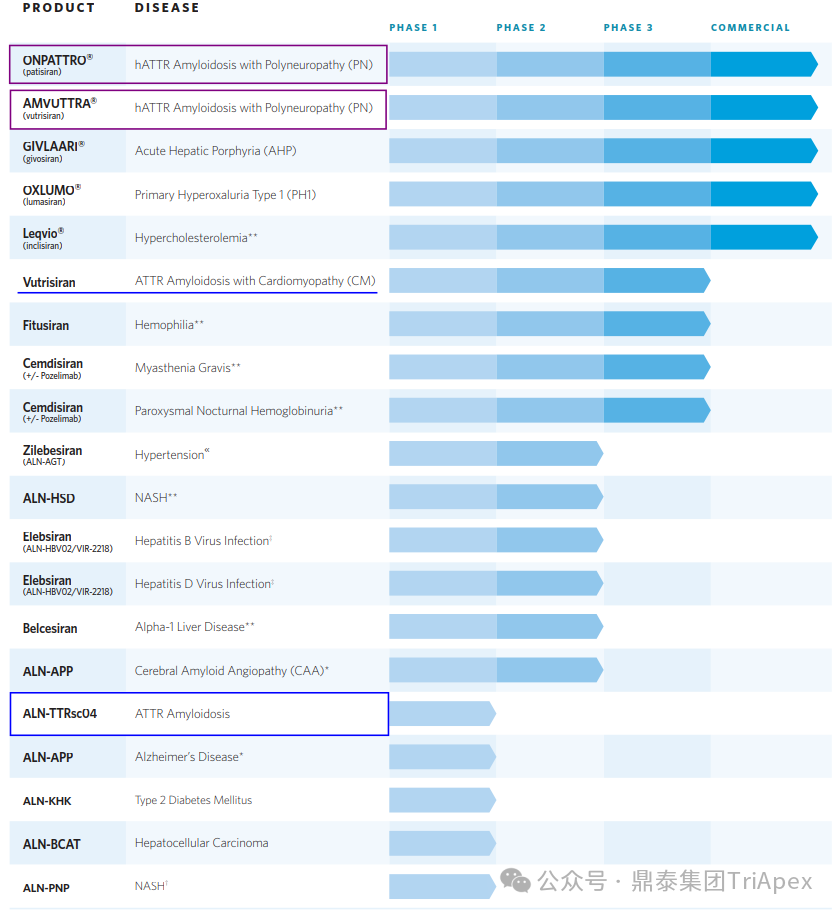
Alnylam 部分产品管线[1]
Patisiran 是 Alnylam 上市的第一款采用 LNP 递送技术的 siRNA 药物,适应症为 hATTR-PN,临床给药途径为 IV,Q3W,推荐剂量为 0.3mg/kg。值得注意的是,Patisiran 用药前需要给予皮质类固醇等来减少与 LNP 相关的炎症反应。Alnylam 在后续的管线开发中放弃了 LNP 递送技术,转而开发 GalNAc 递送技术。在此基础上,Alnylam 迭代开发了 Vutrisiran,是第二款针对 hATTR-PN 的药物,采用了 GalNAc 共轭递送平台,提高了 siRNA 的药效及稳定性。
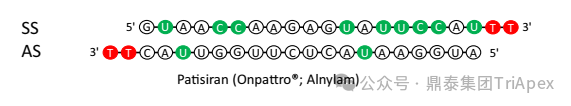
Patisiran 化学结构示意图
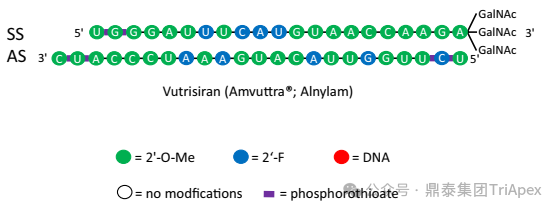
Vutrisiran 化学结构示意图
该产品临床给药途径采用 SC,相对于 Patisiran,给药频率延长至 Q3M,推荐剂量降低为 25 mg。此外,Alnylam 有计划后续将给药频率进一步降低,达到每半年给药一次。Vutrisiran 上市以后,Alnylam 仍针对 ATTR 继续进行迭代产品的开发。如处于临床阶段的 ALN-TTRsc04 采用了更新一代的 IKARIA 平台技术,有望达到更高的给药剂量,实现皮下注射每年给药一次,并将血清 TTR 水平的下调维持在 90% 以上。
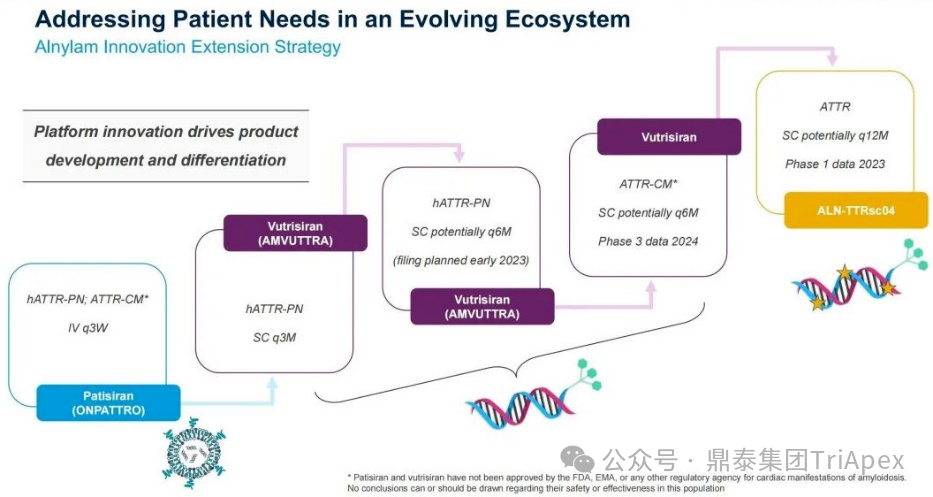
Alnylam 针对 ATTR 疾病领域的技术迭代(Alnylam 公开资料)

Alnylam 针对 ATTR 疾病领域的临床试验布局(Alnylam 公开资料)
1.3 产品上市关键里程碑
2016 年 06 月 07 日,由 Alnylam 在英国开展临床 I 期试验,用于治疗淀粉样变性(NCT02797847)。
2019 年 02 月 14 日,在阿根廷、澳大利亚和比利时等国家和地区开展III期临床试验,用于治疗家族性淀粉样变性和 ATTR-PN(NCT04153149; NCT03759379)。
2022 年 06 月 13 日,Vutrisiran 获得美国食品药品管理局 FDA 批准(NDA215515),由 Alnylam Pharmaceuticals Inc 销售,商品名为 Amvuttra®。
2022 年 09 月 15 日,Vutrisiran 获得欧洲药品管理局 EMA 批准(EMEA/H/C/005852),由 Alnylam Netherlands Bv 销售,商品名为 Amvuttra®。
2022 年 09 月 26 日,Vutrisiran 获得日本医药品与医疗器械局 PMDA 批准(30400AMX00432000),由 Alnylam Japan Kk 销售,商品名为 アムヴトラ皮下注25mgシリンジ®。

Vutrisiran 临床开发路径
Vutrisiran 从首次 IND 申请到获批上市历时约 6 年时间,支持 Vutrisiran 首次 NDA 批准的主要临床试验包含 1 项 Ⅰ 期试验和 1 项 Ⅲ 期临床试验。
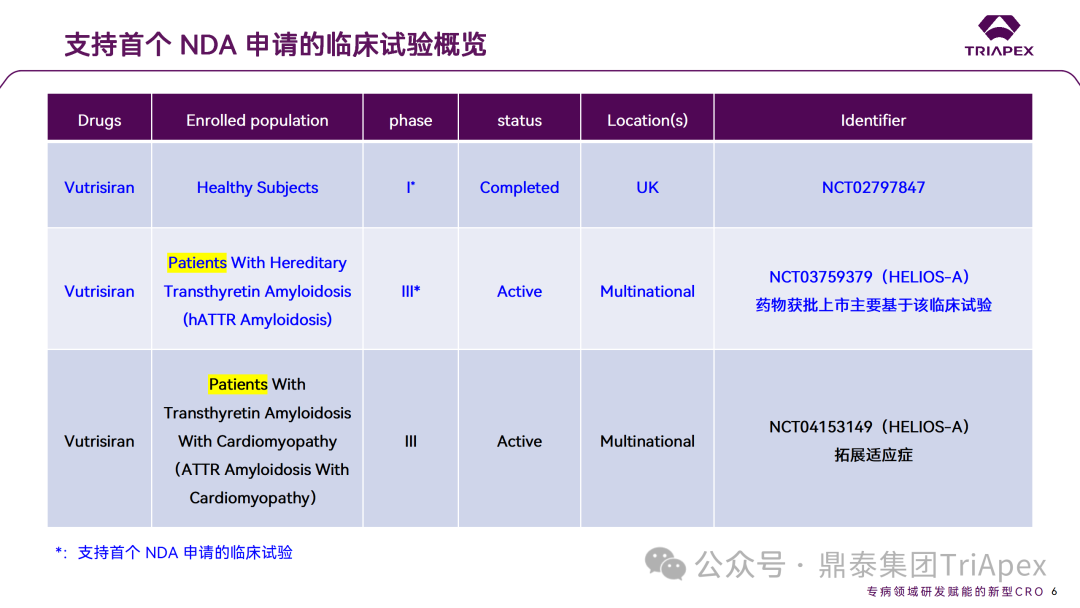
◆ I 期临床试验(NCT02797847)
主要目的是评估 Vutrisiran 在健康受试者中的安全性、耐受性、药代动力学以及药效学特征。
◆ Ⅲ期临床试验(NCT03759379,HELIOS-A)
主要目的是评估 Vutrisiran 对遗传性甲状腺转蛋白淀粉样变性患者的疗效和安全性。在 18 个月的治疗期间,参与者将接受 Vutrisiran 皮下注射(Q3W)或 Patisiran 静脉注射(Q3W)。
该研究使用 APOLLO 研究(NCT01960348,Patisiran 的临床 Ⅲ 期研究,详见第一期)的安慰剂组作为 18 个月治疗期间的外部对照组。在 18 个月的治疗期之后,所有受试者将随机接受每 6 个月一次(Q6M)或在随机治疗延长(RTE)期接受 Q3M 的药物注射治疗。该研究预计 2026 年 10月完成,此次 Vutrisiran 的获批是基于该研究 9 个月的积极结果。
◆ Ⅲ期临床试验(NCT04153149,HELIOS-B)
主要目的是评估 Vutrisiran 25 mg (SC,Q3M) 与安慰剂相比在伴有心肌病的 ATTR 淀粉样变性患者中的疗效和安全性,将用药适应症拓展到心肌病人群中。该研究预计 2026 年底完成。
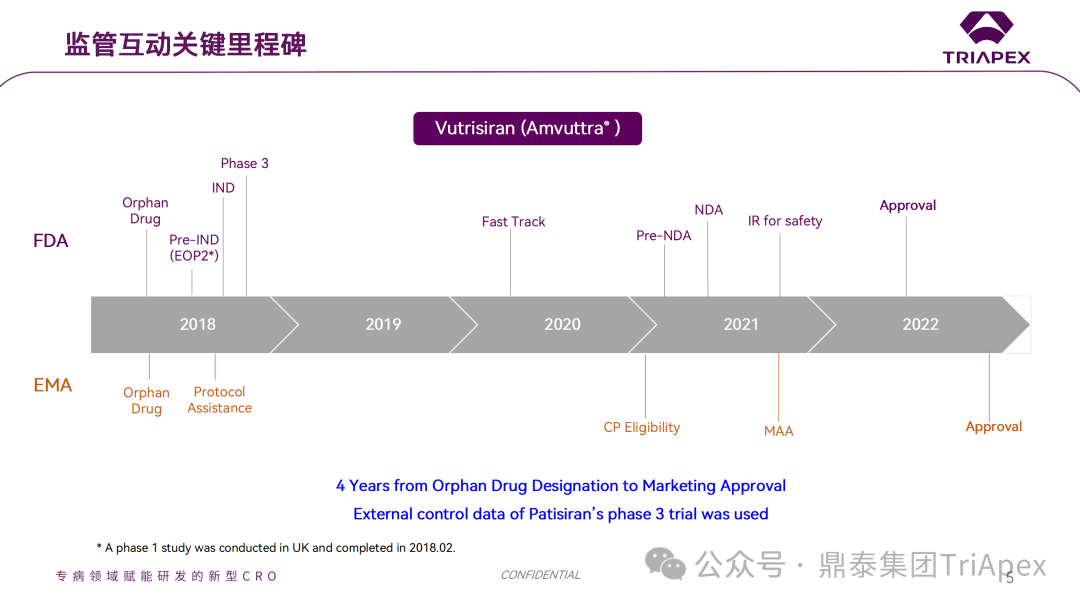
Vutrisiran 临床开发过程中的里程碑事件
2.1 Ⅰ 期临床试验(NCT02797847)
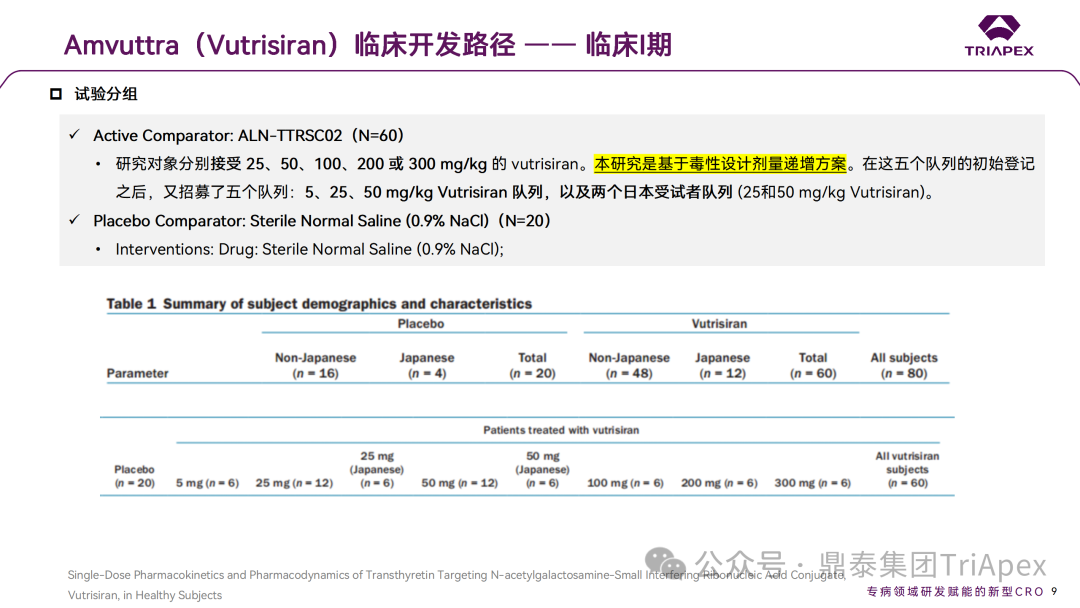
这是一项 Ⅰ 期、随机、单盲、安慰剂对照、单次递增剂量试验,旨在探索药物 Vutrisiran(5-300 mg)在健康志愿者中(n = 80)的安全性、耐受性、药代动力学和药效学特征,同时为 Ⅲ 期临床试验的剂量设计提供依据。

Ⅰ 期临床试验设计
该项 I 期临床试验的方案 (ALN-TTRSC02-001;NCT02797847)经相关独立伦理委员会及机构审查委员会批准。在英国临床研究中心开展试验。健康受试者(年龄 18-65 岁的男性或女性)单次皮下注射给予 Vutrisiran 或安慰剂(生理盐水,0.9%)。
该研究起始剂量的选择主要基于从体内非临床研究(未发表的数据)计算的安全系数[2]。被纳入递增剂量组的受试者分别接受 25、50、100、200 或 300 mg Vutrisiran,研究队列的进展和剂量递增规则基于临床毒性发现(如与 Vutrisiran 相关的具有临床意义的 AE)。在最初招募的 5 个队列的基础上,通过方案修订,又招募了 5 个额外的队列:5、25、50 mg,以及 2 个日本受试者队列 (25 和 50mg),以更好地表征上述剂量下的安全性和有效性,额外的队列开始给药时,先前队列中已显示了可耐受的剂量水平(最高 50mg)。

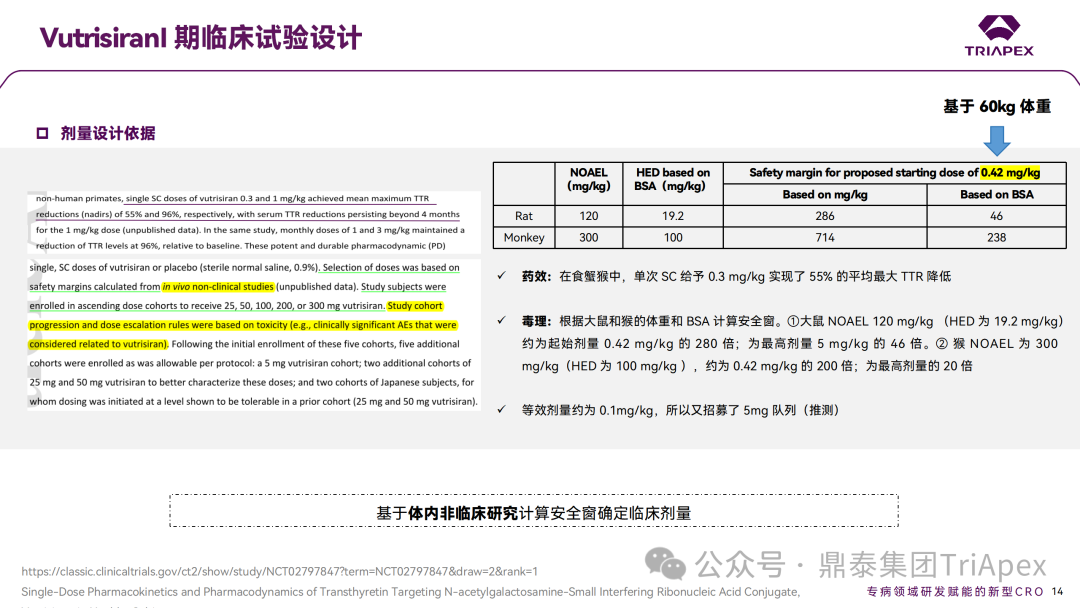
2.1.1 临床Ⅰ期试验起始剂量设计依据
Vutrisiran 的临床 Ⅰ 期试验起始剂量是基于大鼠及食蟹猴的非临床研究,根据 BW 和 BSA 计算安全系数换算而来。在安全窗下推荐的临床起始剂量为 0.42mg/kg,按人体平均体重 60kg 计算,将首次人体试验的起始剂量设定为 25mg。关于后续额外招募的 5mg队列,推测是由于等效剂量约为 0.1mg/kg。
2.1.2 主要研究结果
在健康志愿者中得到的数据表明,Vutrisiran 经皮下注射后能够被迅速吸收,强效并持久地抑制 TTR 蛋白的表达,并且在很宽的剂量范围内具有可接受的安全性。
皮下注射 Vutrisiran 后,10min 即可检测到血浆浓度,3-5h 达到最大浓度。但由于肝脏的摄取,Vutrisiran 的血浆浓度迅速下降,并且在 48h后低于定量下限。但是 Vutrisiran 对 TTR 降低的影响是持久的,单剂量的 Vutrisiran 会导致持续数月的 TTR 表达降低。
在一项非临床研究中,使用其他类似于 Vutrisiran 的 GalNAc-siRNA 的小鼠数据显示,在 SC 给药后 4-24h 观察到肝脏中 siRNA 的峰值浓度,但在给药后 3-7 天观察到最大 RISC 负载的 siRNA 浓度和靶 mRNA 的沉默(与人类相比,大鼠达到最大 PD 效应的时间更短,这归因于大鼠更快的代谢周期和清除,符合异速放大原理)。在这项研究中,健康志愿者经皮下给药后,15-25% 的 Vutrisiran 通过尿液排出体外,其余的药物进入肝脏。这一百分比与大鼠的非临床研究一致,该研究显示有超过 77% 的 GalNAc-siRNA 偶联物位于肝脏中。
降低 TTR 作为 hATTR-PN 治疗策略已通过 Patisiran 得到验证。在健康受试者中,单次 SC Vutrisiran 25mg 可导致持续约 90 天的 TTR 降低 80%。Vutrisiran 25mg Q3M 给药方案预计可实现 88-90% 的 TTR 降低,这与 Patisiran III 期试验(APOLLO)中 Q3M 给药后的 TTR 降低(83-84%)相似。综上所述,预计 Vutrisiran 25mg Q3M 给药方案可带来类似于 APOLLO 研究中观察到的临床获益。试验观察到的积极的药代动力学、药效学和安全性结果支持了 Vutrisiran 的后续临床开发。
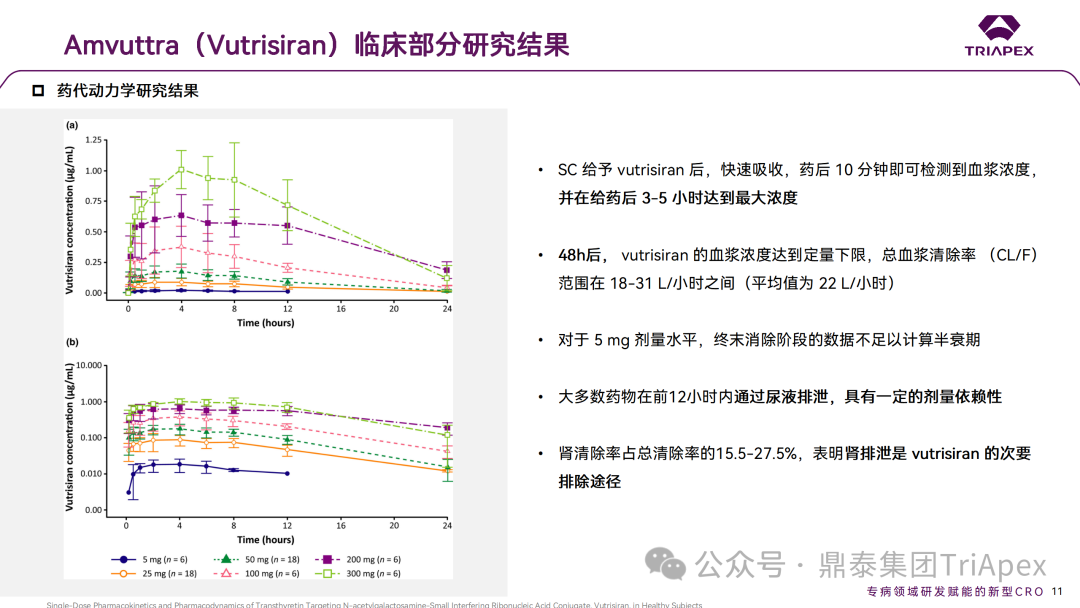
临床药代动力学研究结果

临床药效学研究结果
2.2 Ⅲ 期临床试验(NCT03759379)
HELIOS-A 研究是一项开放标签、国际多中心III期临床试验,旨在评估 Vutrisiran 治疗的有效性和安全性,研究方案和修订得到了审查委员会或独立伦理委员会的批准。入选了 164 例患有多发性神经病的 hATTR 淀粉样变性患者,按照 3:1 的比例随机分为:
Vutrisiran 治疗组(25mg,SC,Q3M),n=122例
Patisiran 治疗组(0.3mg/kg,IV,Q3W),n=44例
来自 APOLLO 研究的外部安慰剂对照组,n=77例
治疗 18 个月。研究的主要终点为与 APOLLO 研究的安慰剂外部对照组比较,Vutrisiran 治疗组9 个月时评估 mNIS+7 评分(改良神经病变损伤评分,量化肌无力、肌肉牵拉反射、感觉丧失和自主神经损伤的评估)较基线的变化值。

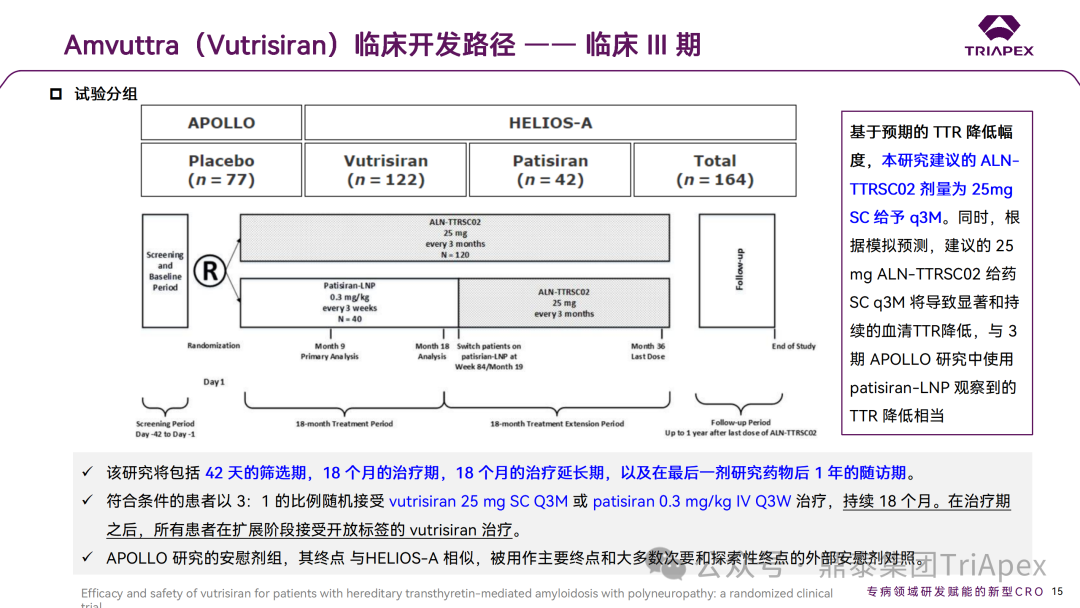
HELIOS-A 试验设计及剂量选择依据

2.2.1 主要研究结果
与安慰剂外部对照组相比,Vutrisiran 治疗在第 9 个月时 mNIS+7 有统计学意义的改善,在第18个月时也观察到了显著的改善;同时,在第 9个月,Vutrisiran 还显着改善了诺福克 QOL-DN 总分,在所有其他次要终点上均观察到 Vutrisiran 治疗的显着改善,包括第 9 个月和第 18 个月的 10-MWT、 mBMI 和 R-ODS。在药效学研究中,Vutrisiran 治疗可导致血清 TTR 水平在 18 个月内快速(≤3 周)和持续的降低,相对于基线可降低 87.6% ,而外部对照组同期为15.7%[3]。
HELIOS-A 的研究结果建立在先前报道的 Patisiran 经验的基础上。本实验中,使用 Vutrisiran Q3M SC 注射液对 TTR 的抑制不逊于 Patisiran Q3W IV 输注,证明了与 GalNac 设计相关的效力和稳定性。
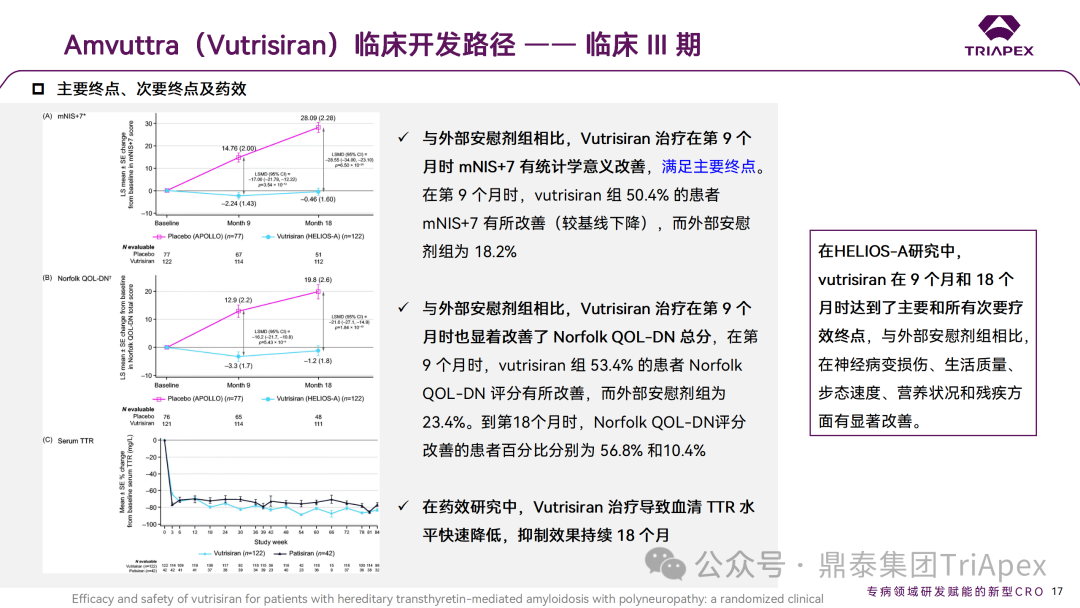
在安全性方面,没有发生与药物相关的停药或死亡事件,最常报告的AE包括关节痛(11%),呼吸困难(7%)和维生素A减少(7%)。在5例患者中报告了注射部位反应(ISRs)(4%),并且都是轻度和短暂的。
综上所述,在 HELIOS-A 研究中,Vutrisiran 在 9 个月和 18 个月时达到了主要和所有次要疗效终点。与安慰剂外部对照组相比,在神经病变损伤、生活质量、步态、营养状况和残疾方面有显著改善,表明疾病表现的逆转,患者可以在这段时间内获得临床益处。Vutrisiran 耐受性良好,并显示出可接受的安全性。除了降低给药频率以外,与 Patisiran 组相比,siRNA 与 GalNAc 的偶联使 Vutrisiran 的皮下给药无需术前用药,同时报告的 ISR AE 更少(4%)。这些都可以减轻患者的治疗负担,提高依从性,更具有临床价值,最大限度发挥长期给药药物治疗的益处。
基于上述III期临床研究(HELIOS-A)9个月的积极结果,FDA 于 2022 年 06 月 14 日批准 Vutrisiran 上市。当然,Vutrisiran 的成功上市,不仅与合理的临床试验设计息息相关,也离不开与监管的高效互动。
监管互动里程碑
3.1 End of phase 2 会议
在 Vutrisiran 的上市历程中,申请人就非临床研究、临床试验设计等问题,多次与监管紧密沟通,确保双方就产品开发和申请策略方面达成一致,避免了不必要的耗时及资源浪费,如 Ⅱ 期临床结束后的 EOP2 会议、上市前的 pre-NDA 会议等。

在 2018 年 7 月 17 日召开的 EOP2 会议上,申请人就 Vutrisiran 的非临床和临床开发计划与 FDA 进行沟通,部分临床相关沟通内容如下:
计划开展的 III 期临床试验将允许轻度肝功能损伤患者、轻度至中度肾损伤患者入组,肝肾功能损伤对 ALN-TTRSC02 PK、PD 和安全性的影响将采用群体 PK/PD 方法进行评估。FDA 是否同意?
FDA答复:
根据提供的信息,ALN-TTRSC02 可能不会受到器官功能损伤的显著影响。FDA 鼓励在研究中纳入中度肝肾功能损伤患者。在 III 期研究中,利用群体 PK/PD 方法来评估内在/外在因素对疗效/安全性的影响是合理的。
在实际临床试验运行过程中,没有针对肝肾损伤患者进行单独的临床试验,在两项临床研究中纳入了轻度至中度的肾损伤患者,在Ⅲ期研究中纳入了中度肝损伤患者。在肝肾损伤患者中,未见 PK 参数的显著差异,后续临床用药无需进行剂量调整;未开展针对重度肝肾损伤患者的研究。
FDA 是否同意针对拟开展的针对 hATTR 淀粉样变性患者的单臂 III 期研究 ALNTTRSC02-002 研究设计,特别是关于外部对照、患者人群选择的纳入标准和研究的持续时间?
FDA答复:
FDA 强烈建议申办方包含一个外部对照(作为罕见病适应症,没有入组安慰剂对照组,采用之前 Patisiran 的对照组作为外部对照更加伦理)来评估 ALN-TTRSC02 与现有疗法相比带来的益处。虽然 FDA 理解申办方基于治疗 9 个月后 Patisiran 观察到的临床益处,但申办方期望观察到 ALN-TTRSC02 临床益处所需的时间可能与 Patisiran 不同。因此,FDA 建议设立 18 个月的观察期,9个月时进行中期疗效分析。
FDA 认为患者人群的纳入标准是可以接受的。
FDA 是否同意 III 期研究 ALN-TTRSC02 所建议的主要终点和其他关键终点?
FDA答复:
主要终点 mNIS+7,是基于临床检查的神经病变损害评分,评分的许多单独组成部分(如神经传导研究),是生物标志物,本身并不代表直接的临床益处,也不是临床益处的直接衡量标准。因此,FDA 建议将 mNIS+7 和 Norfolk QOL-DN(或其他有临床意义的终点)指定为共同主要终点,TTR 生物标志物百分比降低将适合作为次要终点。
综上,申请人在 EOP2 会议中提问了有关Ⅲ期临床试验纳入人群,临床研究设计,主要及次要研究终点等问题,FDA 针对上述问题均给出了监管层面的答复,如增加外部对照组,延长观察时间,设立中期检查点等,但有些建议并未被申请人采纳,比如临床试验主要终点的设计,这可能是基于申请人对自己产品更加独到的理解。
3.2 pre-NDA 会议
在 pre-NDA 会议中, FDA 就申请人拟提交的 clinical data package 是否足以支持 Vutrisiran 用于转甲状腺素运载蛋白淀粉样变性神经病患者治疗的 NDA 审查等问题给出回复。

在 2021 年 3 月 26 日召开的 pre-NDA 会议上,申请人就 Vutrisiran 的 Clinical meeting package 与 FDA 进行沟通,部分相关沟通内容如下:
FDA 是否同意关键的 III 期 HELIOS-A 研究的当前结果和拟提交的临床数据包足以支持 NDA 的审查?
FDA答复:
从表面上看,III 期研究(HELIOS-A)的结果和简报文件中提出的临床数据包有可能支持 Vutrisiran 提交 NDA。在提交 NDA时,将确定数据是否足够。
从简报文件中尚不清楚已获得至少 1 年安全性数据的患者数量。请在提交 NDA 申请时说明将有多少患者已获得至少1年的安全性数据。
此外,与FDA 沟通了关于 NDA data package 准备方面的问题。FDA 给出了共性建议。
综上,在 pre-NDA 会议上,申请人没有询问过多的有关科学性以及试验设计的问题,只是就当前已有数据以及格式咨询 FDA,是否足以支持产品的 NDA 申请,说明申请人对自己的产品在科学设计及数据质量上是有信心的。
免疫原性检测
4.1 Vutrisiran 非临床免疫原性
在非临床研究中采用经验证的 ELISA 法检测食蟹猴血清中抗 Vutrisiran 的 ADA,评价免疫原性。研究结果显示,ADA 的发生率较低,滴度较低,且 ADA 的发生未表现出明显的剂量相关性,申请人认为抗药抗体的存在不会影响食蟹猴体内的药物暴露。这与前几期文章中分享的其他 GalNac-siRNA 的相关研究结果类似。

4.2 Vutrisiran 临床免疫原性
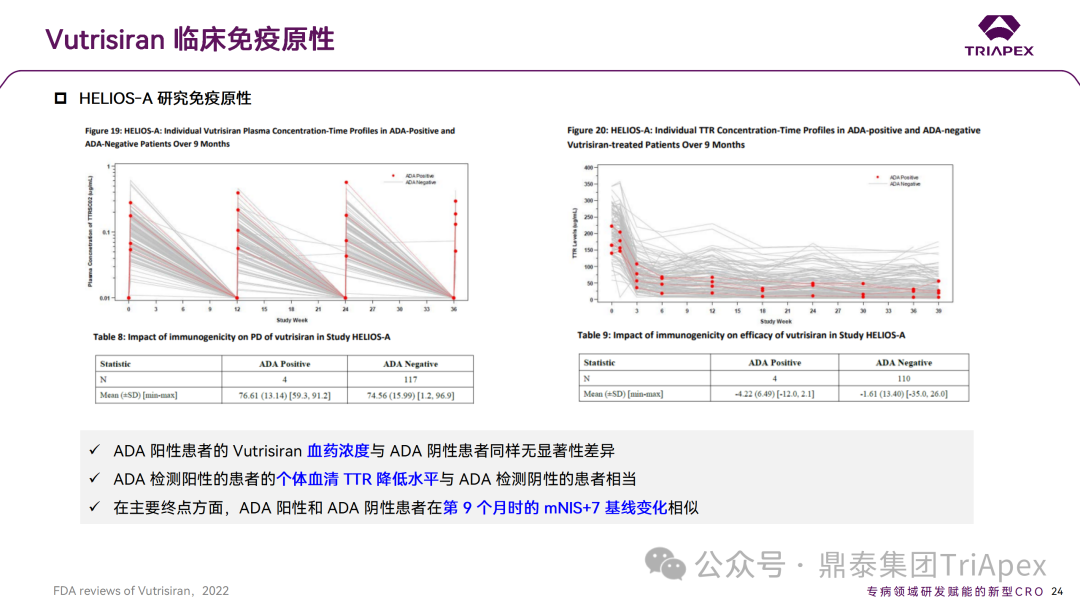
根据 Ⅰ 期试验设计,在给药前(第 -1 天)和给药后第 29 天和第 90 天收集受试者血样用于抗药抗体检测[2]。结果显示,仅有 1 名 (1.7%) 非日本受试者在第 29 天血液检测呈 ADA 阳性,未见有临床意义的显著差异。以上结果提示,Vutrisiran 具有较低的免疫原性,且不会对疗效和安全性产生影响。
在 III 期临床试验(HELIOS-A)中,由药物治疗导致的 ADA 发生率为 2.5%(3/120),且滴度很低(50),患者在后续的随访中检测为阴性。在 ADA 阳性患者和阴性患者的 PK、PD 和疗效无显著差异。ADA 阳性患者的 Vutrisiran 血药浓度与 ADA 阴性患者同样无显著性差异。ADA 阳性患者的个体血清 TTR 降低水平同样与 ADA 阴性患者相当;在主要终点方面,ADA 阳性阴性患者在第 9 个月时的 mNIS+7 基线变化相似。由于较低的发生率和轻微的影响,ADA 的中和活性预计同样不会影响免疫原性评估,因此未对 ADA 的中和活性进行检测[3]。
总体而言,在 2 项临床研究中,Vutrisiran 治疗患者出现 ADA 的发生率较低(2.2%,4/120),滴度很低(50)且是短暂的。同时,ADA 的出现未见时间依赖性,最早发生在第 29 天,最晚发生在第 260 天。在2项研究中,ADA 的存在均未影响 PK、PD 或安全性,在 HELIOS‐A 中未观察到 ADA 对疗效的影响[4]。
非临床与临床试验相关性分析
药物开发过程中,非临床研究和临床研究之间有着密切的关联。非临床研究主要在动物模型中进行,评估药物的安全性、毒性和药代动力学特性,提供药物进入临床试验所需的基本安全性及有效性数据。基于这些数据,临床研究分为多个阶段:I 期临床关注药物的安全性和耐受性,II 期临床评估药物的有效性和进一步的安全性,III 期临床则验证药物在更大人群中的效果和安全性。非临床研究为临床研究提供了科学依据,确保药物开发过程既科学又安全。
5.1 siRNA 药物临床起始剂量与非临床的相关性
FDA 指导原则 Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers(2005) 建议在采用健康志愿者进行 FIH 时,根据 NOAEL 计算起始剂量(MRSD),同时基于药理学活性剂量(PAD)考虑较低的 MRSD 的必要性。
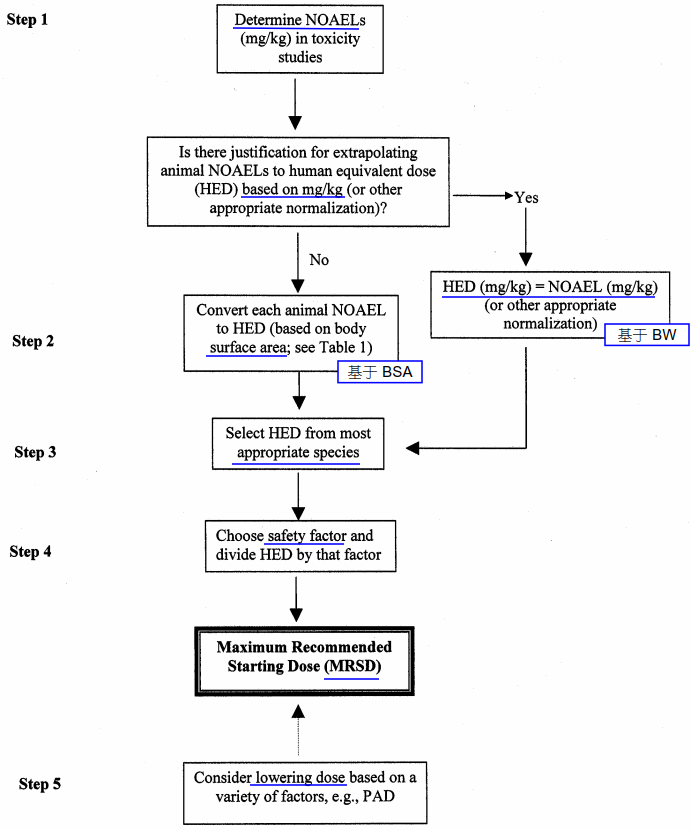
FDA 指导原则中关于健康志愿者作为受试者的 FIH 起始剂量设计依据
基于 Alnylam 的研究经验,包括人体在内的多个种属的寡核苷酸药物 PK 研究表明,将动物剂量外推至 HED 的最合适方法是基于单位体重剂量(mg/kg),而不是单位表面积剂量(mg/m2)或血浆暴露量(AUC 或 Cmax)。寡核苷酸药物的人体等效剂量可以直接从猴外推到人体,按 mg/kg 剂量给药的比例因子为 1.0。
鼎泰团队通过对已上市的 siRNA 的临床起始剂量以及非临床 NOAEL 剂量进行汇总,发现了一些共性和个性的思路。如对于有 Alnylam 公司开发的产品,FIH 起始剂量的计算,综合了非临床药理学研究中的有效剂量以及毒理学研究中最相关种属的 NOAEL 剂量确定起始剂量。此外,我们也注意到,有部分药物在 FIH 起始剂量组开始给药后,入组了更低剂量的受试者人群,推测是直接从动物体内换算而来的人体等效剂量与实际检测结果可能存在差异,或为了探索更广泛剂量下的 PK、PD 特征,重新招募了更低剂量的受试者人群。
以下是已上市的 5 款 siRNA 药物起始剂量设计的依据,其中由 Dicerna 公司研发的 Nedosiran 的思路有所不同,详情敬请关注他山之石系列-6。
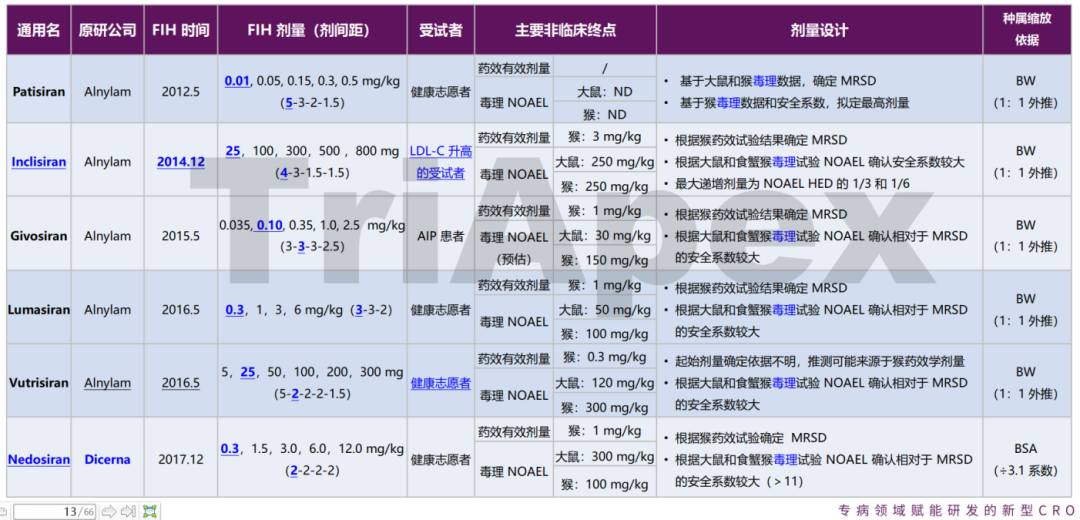
已上市 siRNA 药物 FIH 起始剂量设计依据
5.2 非临床与临床试验的相关性
| 为临床药效学标志物提供参考
食蟹猴体内研究表明,单次给药及多次给药后 Vutrisiran 可剂量依赖性持续降低血清中 TTR 蛋白的水平。在非临床研究中观察到的生物标志物(TTR)的变化,为临床研究中药物剂量设计、给药方案及药效标志物的选择提供了依据,因此在临床试验上,将 TTR 作为主要药效学指标。将 TTR 蛋白作为临床上的药效标志物也得到了之前 Patisiran 研究中的支持。

| 提供剂量设置依据
非临床阶段发现的肝脏毒性对临床试验中起始剂量的设计具有影响,肝脏毒性可能会导致临床不良反应。虽然肝脏毒性属于 siRNA 药物普遍存在的毒性,但在制定临床试验的起始剂量时,需要综合考虑非临床研究中发现的肝脏毒性数据,起始剂量的选择应在保证药物疗效的同时,尽可能降低潜在的肝脏毒性风险。需要基于非临床毒理学研究结果、剂量反应关系以及患者的个体差异来制定,可能需要采取逐步递增的剂量调整策略,以确保药物在安全范围内使用。
| 提示潜在不良反应
通过毒理学数据报告和风险评估,强调肝脏毒性的表现和可能的临床后果。
设计临床试验时应包括肝脏功能的监测指标。制定严格的剂量递增方案和紧密的安全监测计划,及早发现和管理任何与肝脏毒性相关的副作用,有效地评估药物的安全性并保护受试者的健康。
小结
作为寡核苷酸药物领域的头部研发机构,Alnylam 已经积累了丰富的产品上市经验。凭借在技术创新、临床研究策略和药政策略方面驾轻就熟的运用和改进,不断推进新的研发平台和产品迭代,在这种背景下推出了 Vutrisiran。
作为 Alnylam 第二款获得 FDA 批准的 ATTR 领域 siRNA 药物,其上市历程和临床开发路径彰显了对前期 Patisiran 临床数据的合理高效运用,卓越的临床研究策略以及与 FDA 的良好沟通互动的重要性。这些因素对于加快临床转化和推动产品上市至关重要。借助高效的药政策略,Alnylam 与 FDA 就产品上市历程中的关键问题进行了沟通,并获得了 FDA 的认同和支持,表明了其实验设计的合理性以及数据的有效性。同时,利用前期Patisiran的临床数据作为外部对照组,有效证明了迭代产品的安全性与有效性。正是基于前期 Patisiran 的开发经验,Alnylam 在 Ⅰ 期研究初步验证安全性和有效性之后,敢于直接扩大受试者样本量开展 III 期临时研究,从而加快了产品的上市进程。这些策略和做法也为后续 siRNA 药物的研发提供了宝贵的经验和启示。
Alnylam 从未止步,针对 ATTR 疾病领域展开了更加深远的布局,期待为更多患者带来更多的治疗选择和希望。2024年6月24日,Alnylam 宣布 Vutrisiran 用于治疗 ATTR 淀粉样变性心肌病(ATTR-CM)的III期 HELIOS-B 研究达到了主要终点。结果显示,在双盲期间总人群(N=654)和单药治疗人群(N=395)的全因死亡率和复发性心血管事件的发生率显著降低,所有次要终点均获得统计学显著改善。HELIOS-B 研究表明,Vutrisiran改 善了所有 ATTR 心肌病患者组的心血管状况,包括生存、功能和生活质量,且安全性可控。Alnylam 计划在 2024 年下半年向全球监管机构提交补充上市申请,包括使用优先审评券[5]向 FDA 提交补充新药申请(sNDA)。
综上,Vutrisiran的成功再次印证了与监管机构的沟通、优化临床试验设计以及充分利用非临床研究结果和以往临床经验的重要性,这些策略为后续 siRNA 药物的研发提供了重要经验和启示。Alnylam 正在更多疾病领域进行更深远的布局,期待为更多患者带来治疗选择和希望。
下期,鼎泰团队将分享他山之石系列的最后一期 —— Nedosiran,传承和差异化并存,敬请期待。
参考资料:
[1] Alnylam官网:https://www.alnylam.com/alnylam-rnai-pipeline
[2] Habtemariam BA, Karsten V, Attarwala H, et al. Single-Dose Pharmacokinetics and Pharmacodynamics of Transthyretin Targeting N-acetylgalactosamine-Small Interfering Ribonucleic Acid Conjugate, Vutrisiran, in Healthy Subjects. Clin Pharmacol Ther. 2021;109(2):372-382.
[3] Adams D, Tournev IL, Taylor MS, et al. Efficacy and safety of Vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid. 2023;30(1):1-9.
[4] FDA reviews of Vutrisiran. 2022
[5] Alnylam官网信息:https://investors.alnylam.com/press-release?id=28321







 法规政策
法规政策 项目管理
项目管理


 Global
Global




 苏公网安备32011202001081
苏公网安备32011202001081