


上一期,鼎泰集团对多个已上市ADC药物(小分子毒素为微管抑制剂)的非临床研究路径进行了汇总和复盘(点击查看)。本期我们将分享已上市的以DNA拓扑异构酶1抑制剂为Payload 的ADC药物(TOP1-ADC) Enhertu(靶向HER2, Payload为DXd) 和 Trodelvy(靶向Trop2, Payload为SN-38) 的上市历程和分阶段支持IND和BLA的非临床研究内容。 期待通过本期分享,加深对相关指南的深度理解和具体产品研究方案的精准把握,并期待与广大业内同行一起交流、探讨,共同推动ADC产品的开发。如有不准确之处,欢迎同行提出宝贵意见,我们会持续改进。
喜树碱来源的TOP1,如拓扑替康(Topotecan)、伊立替康(Irinotecan)、贝洛替康(Belotecan)已被批准用于治疗非小细胞肺癌、卵巢癌和结直肠癌[1]。Deruxtecan(DXd+linker)、Exatecan、SN-38(Irinotecan的活性代谢产物) 尚未单独批准上市,常作为喜树碱类Payload用于ADC药物的开发。
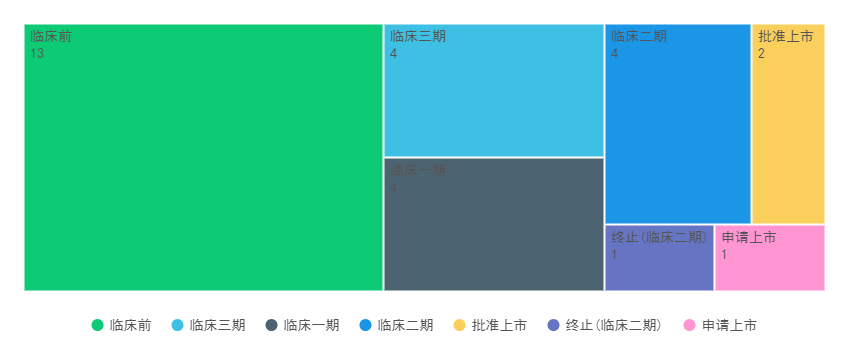
喜树碱及其衍生物的化学结构[1] (A) Camptothecin(CPT), (B)Topotecan, (C)Irinotecan(CPT-11), (D)SN-38, (E) Belotecan, (F)Exatecan(DX-895), (G)Deruxtecan(DX-8951 (DXd)+ maleimide-GGFG peptide linker) TOP1-ADC由于具有独特作用机制的细胞毒作用、高DAR、强的旁观者和稳定且能被特异性切割的连接子,已成为ADC药物开发的重点方向。目前,全球范围内已有2个以TOP1抑制剂为Payload的ADC药物(TOP1-ADC)上市;至少1个TOP1-ADC提交了上市申请;多个TOP1-ADC处于不同阶段的临床试验中。 全球TOP1-ADC的临床研发进展 (鼎泰集团根据药渡数据整理,截至2023.12.28)
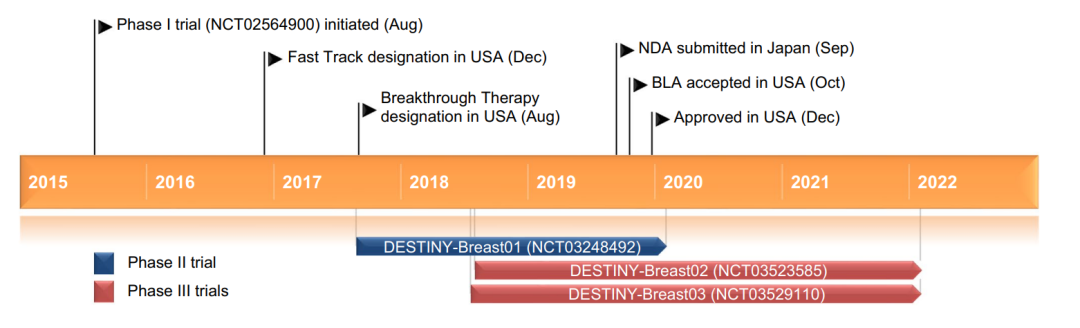
Enhertu®(fam-trastuzumab deruxtecan-nxki) 非临床研究内容 由抗HER2单抗(IgG1)通过可切割连接子GGFG(MAAA-1181a)与拓扑异构酶1抑制剂DXd连接组成。 DAR为8,分子量未披露。 适应症:首次申请获批的适应症为既往接受过基于抗HER2方案治疗、不可切除或转移性HER2阳性乳腺癌成年患者;后续获批适应症包括:不可切除或转移性HER2低表达乳腺癌成年患者;HER2突变、不可切除或转移性NSCLC成年患者;既往接受过以曲妥珠单抗为基础的治疗方案的局部晚期或转移性HER2阳性胃癌或胃-食管交界腺癌成年患者。 用法用量:治疗乳腺癌、肺癌的推荐剂量为5.4 mg/kg/次,治疗胃癌的推荐剂量为6.4 mg/kg/次;所有适应症给药方案均为静脉输注,21天/次,直至疾病进展或出现不可接受的毒性。 Enhertu®(fam-trastuzumab deruxtecan-nxki) 化学结构,图源FDA审评报告 Enhertu®(fam-trastuzumab deruxtecan-nxki) 开发的关键里程碑[2] 2016年1月27日,向FDA提交IND 127553以支持启动I期临床试验(DS8201-A-J101)。 2016年11月10日,获快速通道资格认定(Fast Track Designation) 用于治疗经HER2靶向治疗(包括T-DM1)后进展的HER2阳性、不可切除和/或转移性乳腺癌患者。 2017年8月25日,获突破性疗法资格认定(BTD) 用于HER2阳性、局部晚期或转移性乳腺癌患者的治疗,这些患者已接受曲妥珠单抗和帕妥珠单抗治疗,并在T-DM1后出现疾病进展。 2019年10月17日, 获优先审评资格认定(Priority Review) 用于治疗HER2阳性转移性乳腺癌患者。 2019年12月20日,FDA加速批准(AcceleratedApproval) ENHERTU®, 用于接受过两种及以上抗HER2治疗、不可切除或转移性HER2阳性乳腺癌患者。该上市申请基于如下关键性临床试验: 在一项多中心、单臂试验DESTINY-Breast01(NCT03248492) 中,招募了 184 名既往接受过两种及以上抗 HER2 治疗的 HER2 阳性、不可切除和/或转移性乳腺癌女性患者。以5.4 mg/kg每3周静脉输注一次,直至出现不可接受的毒性或疾病进展。主要疗效终点指标采用 RECIST 1.1 标准判定的ORR和DOR。ORR为60.3%(95% CI:52.9,67.4),CR为4.3%,PR为56%;DOR为14.8个月(95% CI:13.8,16.9)。 分阶段支持该产品IND和BLA的非临床研究内容如下: 备注: 蓝色字体:支持首次人体试验(FIH) IND 申请的非临床研究 黑色字体:支持后续 IND 和 BLA 申请的非临床研究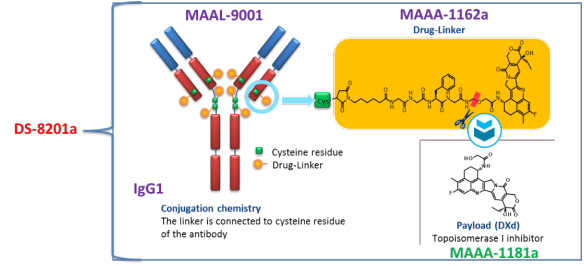

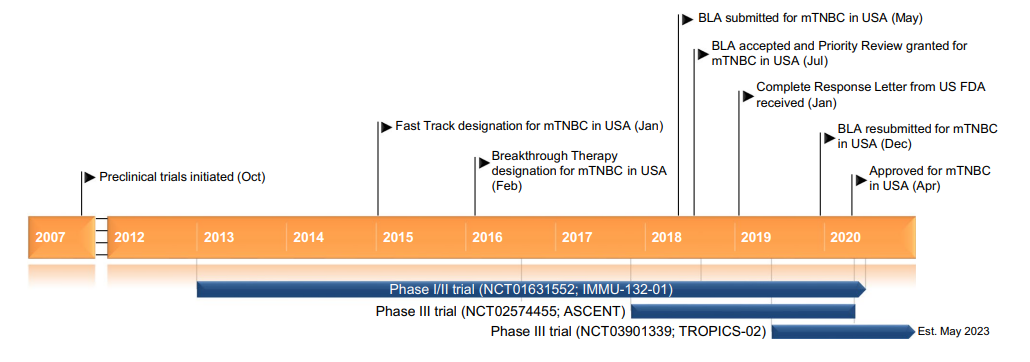
Trodelvy®(sacituzumab govitecan-hziy) 非临床研究内容 由抗Trop2单抗(IgG1)通过可切割连接子CL2A 与拓扑异构酶1抑制剂SN-38连接组成。 DAR:约7.6,分子量未披露。 连接子:CL2A,可水解。 适应症:首次获批适应症为用于治疗既往接受过至少两种治疗的不可切除的局部晚期或转移性三阴性乳腺癌(TNBC)成年患者,包括至少一种局部晚期或转移性疾病的既往治疗;后续批准的适应症包括:不可切除的局部晚期或转移性HR阳性、HER2阴性乳腺癌,患者需接受过内分泌治疗和至少两种其他转移性全身治疗;局部晚期或转移性尿路上皮癌(mUC),患者先前接受过含铂化疗和 PD-1 或 PD-L1 抑制剂。 用法用量:静脉输注,推荐剂量为 10 mg/kg,每周1次,连续21天治疗周期的第1天和第8天,直到疾病进展或不可接受的毒性。 Trodelvy®(sacituzumab govitecan-hziy) 化学结构,图源药渡 Trodelvy®(sacituzumab govitecan-hziy) 开发的关键里程碑[3] 2012年6月1日,评估sacituzumab (IMMU-132, hRS7-SN38) 治疗晚期上皮恶性肿瘤 (IND 115621) 的首次IND申请。 2014年12月22日,获快速通道认定用于既往至少接受过2线治疗的TNBC患者。 2016年2月4日,根据IND 122694获得突破性疗法认定用于既往接受过至少2线治疗的治疗复发/难治性、转移性TNBC患者。 2020年4月7日,获快速通道认定用于转移性尿路上皮癌。 2020年4月22日,FDA批准Trodelvy(sacituzumab govitecan-hziy) 用于既往至少接受过2线治疗的转移性TNBC: 根据一项针对108名转移性TNBC患者的临床试验结果批准了Trodelvy,主要终点为ORR。结果显示,接受治疗的患者ORR为33.3%,DOR为7.7个月。在对Trodelvy有响应的患者中,55.6%维持了6个月或更长时间,16.7%维持了12个月或更长时间。 分阶段支持该产品IND和BLA的非临床研究内容如下: 备注: 蓝色字体:支持首次人体试验(FIH) IND 申请的非临床研究 黑色字体:支持后续 IND 和 BLA 申请的非临床研究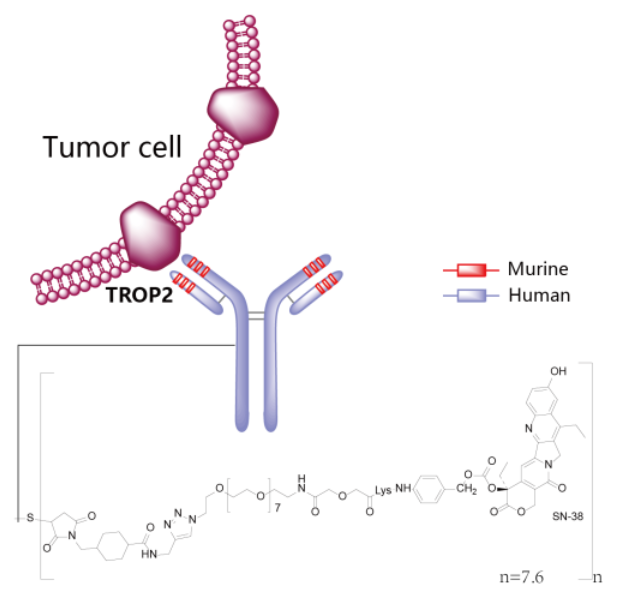

参考资料: [1] Han S, Lim KS, Blackburn BJ, Yun J, Putnam CW, Bull DA, Won YW. The Potential of Topoisomerase Inhibitor-Based Antibody-Drug Conjugates. Pharmaceutics. 2022 Aug 16;14(8):1707. doi: 10.3390/pharmaceutics14081707. PMID: 36015333; PMCID: PMC9413092. [2] Keam SJ. Trastuzumab Deruxtecan: First Approval. Drugs. 2020 Apr;80(5):501-508. doi: 10.1007/s40265-020-01281-4. PMID: 32144719. [3] Syed YY. Sacituzumab Govitecan: First Approval. Drugs. 2020 Jul;80(10):1019-1025. doi: 10.1007/s40265-020-01337-5. PMID: 32529410; PMCID: PMC7288263. 供稿:鼎泰集团转化科学和药政策略部







 法规政策
法规政策 项目管理
项目管理


 Global
Global



 苏公网安备32011202001081
苏公网安备32011202001081