


即从巴峡穿巫峡,便下襄阳向洛阳。
上期文章,鼎泰集团对第一款 siRNA 药物 Patisiran(Onpattro®)的上市历程及临床开发路径进行了复盘分析(点击查看原文),为我们清晰、高效地呈现了一款 siRNA 产品如何从非临床向临床转化,以及借助无缝衔接的临床研发策略在 IND 后的短短 6 年内获批上市的经典案例。该分享受到了同行的高度关注和认可,也鼓舞我们对后续产品进行更进一步的调研、总结和提高。 第2款已上市 siRNA 药物—— Givosiran(Givlaari®) 的上市历程及临床开发路径则更加惊艳。本期内容在第1期分享的基础上增加了更多与临床转化有关的内容,以期为 siRNA 药物如何运用临床前研究结果指导临床开发,以及全局视角的研发提供参考和启发。 Givosiran 的上市历程
Givosiran是由 Alnylam Pharmaceuticals 开发的首个 GalNAc偶联的siRNA 药物(GalNac-siRNA),能够特异性递送至肝脏细胞并靶向降解编码ALAS1蛋白的 mRNA,从而降低神经毒性前体 δ -氨基乙酰丙酸(ALA)和胆色素原(PBG)的水平,减少疾病发作[1]。 Givosiran作用机理示意图 2015年8月13日,Givosiran 提交IND。2019年11月20日,在提交NDA后的不足4个月(比预期的PUDUFA时间提前了近3个月)内即获得FDA批准用于治疗成人急性肝卟啉症(AHP)。此后,先后于2020年3月2日和2021年6月23日获得EMA和PMDA批准上市。该适应症的批准是基于关键性Ⅲ 期临床试验(NCT03338816,代号ENVISION)的积极结果。 Givosiran的成功上市不仅为AHP患者带来了新的治疗选择,也为RNAi技术在治疗罕见疾病领域的应用提供了新的范例。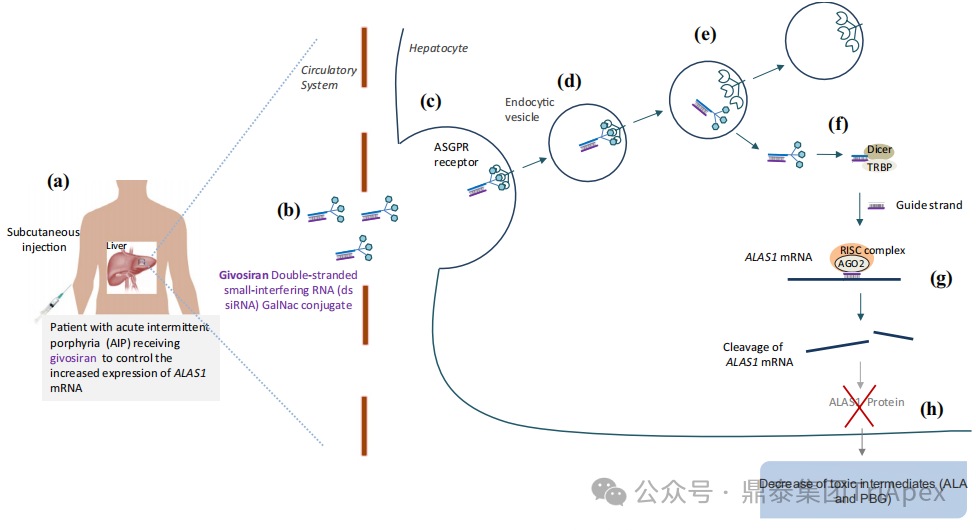
急性肝卟啉症(AHP)
AHP 是由于血红素生物合成途径中第3个[表现为急性间歇性肝卟啉症(AIP)]、第6个[家族性卟啉原卟啉酸尿症(HCP)]或第7个[变异卟啉病(VP)]酶的常染色体显性失活,或在非常罕见的情况下,由常染色体隐性遗传导致的一种罕见、严重、可能危及生命的遗传性疾病。 这些染色体的异常变化导致 ALA 和 PBG 的积累,进而导致急性卟啉症发作,主要表现为脑脊髓与交感神经系统的症状,如腹痛(主要症状)、急性神经病变等。大多数有症状的患者在其一生中很少发作,然而,也有8 %的患者会出现病情加重,反复发作(≥ 4次/年)。最常见的急性肝卟啉症是 AIP,其患病率估计为5.4 /百万人。 静脉注射血红素是当时欧盟和美国推荐的用于AHP急性发作的治疗方案。然而,使用血红素治疗还有诸多限制,除了在水溶液中不稳定外,还包括重复给药可能引起的潜在毒性,如静脉血栓性静脉炎、速效耐药性、凝血异常和继发性铁负荷等。因此,迫切需要更加安全、有效的治疗方案来应对急性肝卟啉症的发作并减少长期并发症。
临床开发路径
无缝衔接、驾轻就熟的临床开发路径是Givosiran成功上市的重要支撑。1项Ⅰ期剂量/给药频率探索试验、1项无缝衔接的Ⅰ/Ⅱ 开放标签扩展试验(OLE)和1项Ⅲ期关键性临床试验为Givosiran的NDA申请和获批上市提供了充分的临床数据支持。 此外,上述临床试验的高效推进离不开该产品非临床研究结果的支持,以及从先驱产品Patisiran开发过程中积累的宝贵经验。 Givosiran临床前和临床开发路径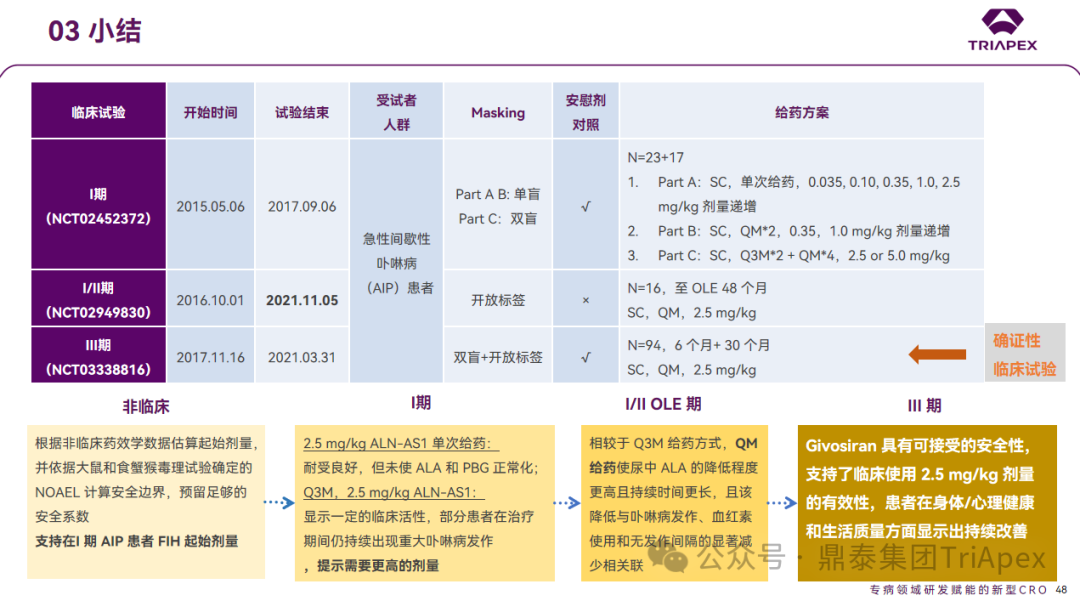
1 Ⅰ 期临床试验(NCT02452372,跨地区多中心) 2015年,Givosiran 的首次人体试验(FIH)启动。这是1项多中心、随机、安慰剂对照的剂量递增试验,旨在评估 Givosiran 在 AIP 患者中的安全性、耐受性及药代动力学/药效动力学特征。

试验分3部分进行,共入组了40人,均为皮下注射给药: A和B部分共入组23人,C部分入组17人。A部分(单次给药,0.035、0.10、0.35、1.0、2.5 mg/kg,42天)和B部分(QM,0.35、1.0 mg/kg,70天)为单盲试验,受试者为尿ALA和PBG水平升高但近期未发作(定义为在给药前6个月内没有发作)的 AIP 患者; C部分(Q3M+QM,2.5、5.0 mg/kg,168天)为双盲试验,受试者为反复发作的 AIP 患者。 主要终点为给药后发生 AEs、SAEs及中断用药的受试者比例;次要终点为Givosiran 药代动力学参数(Cmax、tmax、t1/2、AUC)以及对尿中ALA和PBG水平的影响。 起始剂量设计是从非临床研究向临床转化的第一步。本试验中起始剂量主要依据: ◆ 非临床药效学数据: (1)大鼠AIP模型中,Givosiran以0.3、1或3 mg/kg(QW*4)皮下注射给药后,可剂量依赖性降低 ALAS1 mRNA 水平,同时伴随着尿液中ALA和PBG水平的下降; (2)小鼠AIP模型中,Givosiran在 1 mg/kg 剂量下,血清 ALA 水平降低80% 和血清 PBG水平降低 68; (3)食蟹猴模型中,Givosiran 以1 mg/kg 皮下注射给药后,ALAS1 mRNA 水平仅降低20%。根据前期项目经验,假设从NHP到人按剂量比1:1 转化,预期Givosiran 在0.1mg/kg剂量下不会在人体内产生药理学活性。 Givosiran 非临床药效部分数据(请见往期内容) ◆ 非临床毒理学数据: 根据大鼠和食蟹猴重复给药毒理学试验中确定的 NOAEL 计算安全系数,最终采信了相对保守的基于 BSA 估算的安全系数。 基于以上非临床研究结果,支持在 Ⅰ 期 AIP 患者研究中 FIH 起始剂量为 0.10 mg/kg。 研究结果显示,对反复发作的 AIP 患者,经 QM 给药后的主要AEs均为轻度 AEs;可降低 ALAS1 mRNA 水平,ALA和PBG的水平接近正常,发作率低于安慰剂组[2]。




Ⅰ期试验主要研究结果示意图
2 Ⅰ/Ⅱ 期临床试验(NCT02949830) 2016年,启动了 Ⅰ/Ⅱ 期临床试验。这是1项多中心、开放标签扩展(OLE)研究,旨在评估皮下注射 Givosiran 的长期安全性和临床活性,受试者为16名已完成前述I期临床试验(NCT02452372)C部分的 AIP 患者。主要终点为截至49个月时的 AEs 发生率;次要终点为截至48个月时尿液中 ALA 和 PBG 水平的变化、卟啉病发作的频率和特点以及截至49个月时血红素给药情况的变化。 该试验给药剂量为2.5 mg/kg,QM皮下注射。根据 Ⅰ 期临床试验结果,Givosiran 2.5 mg/kg 单次给药耐受性良好,但没有完全使 ALA 和 PBG 水平正常化;Givosiran 2.5 mg/kg Q3M给药显示一定的临床活性,但部分患者在治疗期间仍持续出现严重卟啉病发作,这提示需要更高的给药剂量。 研究结果显示,Givosiran 2.5 mg/kg,QM给药安全性良好,大多数AEs为轻度至中度;长期使用 Givosiran 使 ALA 和 PBG 水平持续降低,没有卟啉症发作和不需要使用血红素的患者比例随着时间推移增加;安全性和有效性结果与下文所述的 Ⅲ 期临床试验(ENVISION)的结果一致[3]。



Ⅰ/Ⅱ期试验主要研究结果示意图 3 Ⅲ 期临床试验(NCT03338816,代号ENVISION) 2017年,1项随机、双盲、安慰剂对照的6个月多中心 Ⅲ 期临床试验启动(双盲期后进行30个月开放标签扩展试验)。该研究共完成36个月的数据分析,旨在评估 Givosiran 治疗急性肝卟啉病患者的疗效和安全性,受试者共94人。ENVISION 试验在18个国家/地区的36个研究地点进行研究,是AHP进行的最大规模的治疗研究。 ENVISION试验中给药剂量为2.5 mg/kg(QM),剂量设计主要基于对前期试验结果的考量:相较于 Q3M,QM 给药使尿中 ALA 的降低程度更高且持续时间更长,且该降低与卟啉病发作、血红素使用和无发作间隔的显著减少相关联。主要终点为需要住院、紧急医疗就诊或在家接受静脉血红素治疗的卟啉病发作的年化发作率(AAR);次要终点为3、6个月时尿ALA水平,6个月时尿PBG水平、血红素使用的比例、简明疼痛量表(BPI-SF)及SF-12健康调查量表等。 ENVISION试验获得了支持Givosiran用于AHP治疗的有效性证据[4]: 主要终点:Givosiran治疗(<−36个月)导致急性间歇性卟啉病症状持续改善,没有发作的患者比例增加。 次要终点:尿中 ALA/PBG 水平在整个治疗期持续降低;随着时间的推移,没有发作或不需要使用血红素的患者比例增加;Givosiran 具有良好的安全性,且患者在身体/心理健康和生活质量方面显示出持续改善。

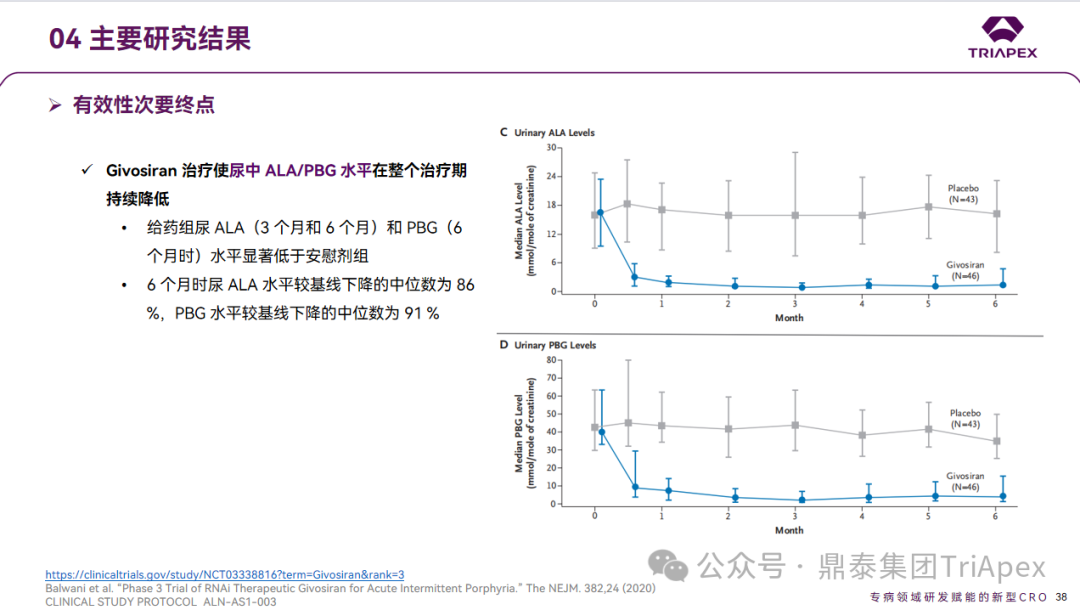

ENVISION试验主要研究结果示意图
监管里程碑
除了成功的临床开发计划外,Givosiran惊人的上市历程还受益于与监管机构的有效沟通。 基于Ⅰ期临床试验结果,Alnylam与FDA 于2017年6月14日召开 Pre-Phase 3会议(此时I/II 期 OLE 试验已启动),主要就拟开展的 Ⅲ 期试验设计征求 FDA 的建议。FDA 认为申请人提出的剂量设计以及主要/次要终点是合理的,建议考虑将接受 Givosiran 治疗的 AHP 患者的患者报告结局(Patient Reported Outcomes,PRO)的改善作为一个关键终点。通过本次沟通交流获得的建议,使ENVISION 试验的有效性结果更快地获得了FDA的认可。 Pre-Phase 3 会议部分沟通问题和建议 此外,在从IND和NDA的过程中,还有一系列其他的监管互动,如ODD、BTD、B类会议、优先审评等。

非临床与临床试验相关性分析
在非临床药效学研究中,对于Givosiran作用机制相关的药效学 Biomarker 的变化规律进行了探索,这些研究不但支持了FIH起始剂量的设计,也为临床试验中药效动力学检测指标和监测时间的确立提供了依据。 | 非临床药效学研究 采用啮齿类和非人灵长类动物模型进行的AHP非临床研究中:以0.3、1或3 mg/kg(QW*4)剂量大鼠AIP模型皮下给药后,与单次给药类似,对 ALAS1 mRNA 的抑制具有剂量依赖性,同时伴随着尿液中ALA和PBG水平的相应下降; 以 2.5、5 mg/kg(QW*8)剂量食蟹猴皮下注射Givosiran后,血清中 ALAS1 mRNA 水平最大减少约 80%。这些试验不但考察了Givosiran用于AHP治疗的作用机制,也为临床试验中合理的可用于监测和提示预期疗效的Biomarker的确立提供了参考。 | 临床试验结果 依赖于对Givosiran作用机制的理解以及非临床研究结果,在I期、I/II临床OLE期和 III期临床试验中均考察了给药后ALAS1 mRNA、ALA及PBG水平等药效学标志物的变化,并将其作为剂量和给药频率选择的依据。结果显示,Givosiran 2.5 mg/kg,QM 给药,能够降低肝脏中ALAS1的水平,同时尿中ALA和PBG的水平也降低到了正常范围。 该结果与临床前药效学研究结果具有较好的相关性,从侧面提示了非临床药效学研究对于提示临床试验设计的重要性。 肝脏毒性是非临床和临床对应关联的毒性表现,可在临床阶段进行密切监控,全面评估药物的安全性和风险:非临床结果显示肝细胞单细胞坏死、肝细胞嗜碱性颗粒等;临床中也有一定肝毒性结果。 对于寡核苷酸药物,非临床研究中发现的毒性与临床试验中出现的 AE 具有较高的关联性。Givosiran非临床结果显示肝细胞单细胞坏死、肝细胞嗜碱性颗粒等,临床中则可见转氨酶升高和肝脏毒性,并在说明书中给予警示。上述关联性提示,非临床研究对于评估临床安全性和指导风险控制计划的制定具有重要价值。 综上所述,非临床研究为临床试验设计提供了依据;科学的剂量和给药方案探索、无缝衔接的临床研究规划、合理的III期临床试验设计是Givosiran快速获得监管机构上市批准的重要保障。此外,基于稳健的前期数据,与监管机构进行有效的沟通也是必不可少的药政策略。Givosiran的上市历程和临床开发路径的复盘分析为siRNA药物的研发提供了宝贵的经验和启示。

参考资料: [1] de Paula Brandão et al. “Leading RNA Interference Therapeutics Part 2: Silencing Delta-Aminolevulinic Acid Synthase 1, with a Focus on Givosiran.” Molecular diagnosis & therapy vol. 24,1 (2020): 61-68. [2] Sardh E et al. Phase 1 Trial of an RNA Interference Therapy for Acute Intermittent Porphyria. N Engl J Med. 2019;380(6):549-558. [3] Final Results from a Phase 1/2, 48-Month, Open-Label Extension Study of Givosiran in Patients with Acute Intermittent Porphyria. European Association for the Study of the Liver (EASL) Congress 2023. [4] Balwani M et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N Engl J Med. 2020;382(24):2289-2301.







 法规政策
法规政策 项目管理
项目管理


 Global
Global



 苏公网安备32011202001081
苏公网安备32011202001081