


随着药物研发的推进,致癌性试验成为支持部分药物上市申请的非临床安全性评价的重要内容之一。鼎泰团队对药物致癌性试验的必要性、致癌性试验的设计和数据分析以及致癌性试验相关的资源可及性进行了充分的调研。本文通过回顾《ICH S系列指导原则宣讲会》,及FDA对减少致癌试验动物指南的解读,提出鼎泰团队对药物致癌性试验的思考和建议,力求为国内同行、新药研发企业和审评机构提供参考。
01
CDE关于ICH S1/S1B(R1)
药物致癌性试验指导原则专家解读
关于CDE于2022年11月17日《ICH S系列指导原则宣讲会》(线上会议,国家药品监督管理局ICH工作办公室主办)中,由孙涛老师对S1/S1B(R1):药物致癌性试验进行解读,整体从S1、S1B(R1)修订情况及S1实施思考与关注问题等方面做了介绍。
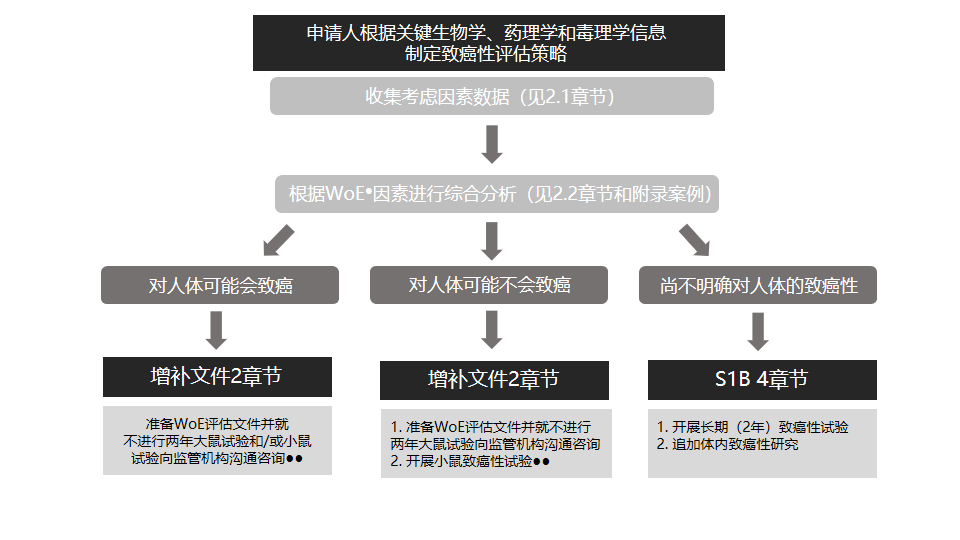
S1B(R1)内容摘要,制定致癌性评估策略和确定2年大鼠致癌性试验附加价值的关键步骤和选项的流程图。(来源:ICH S系列指导原则宣讲会)
针对S1B(R1)提出如下相关考虑:
1、当WoE综合评估支持提出“进行两年致癌性试验不会为人体致癌性风险评估提供更多有价值的信息”的结论时,申请人应按照当地既定的监管沟通程序,向相应的监管机构咨询意见;
2、当申请人决定根据ICH S1B进行2年大鼠试验时,无需征求监管机构的同意:
a. 采用小鼠进行的致癌性试验,无论是依据ICH S1B在标准品系小鼠中进行的2年试验还是在转基因模型中进行的短期试验,仍然是致癌性评估计划中推荐的组成部分;
b. 有些情况下,可能不适合进行小鼠致癌性试验。例如:
● 当WoE评估强烈表明对人体无致癌性风险,且数据显示,相对于人体暴露量,在小鼠体内只能达到亚治疗剂量和无药理学活性的药物水平。
● 当WoE评估表明一种化合物对人体可能有致癌性。
02
FDA专家对减少致癌试验动物
使用最新指南的解读
2022年2月,FDA改变过去首先发表的指南草案的做法, 直接向制药工业发出终版的指南,要求业界在COVID-19期间尽量减少使用非人类灵长类(NHP)开展非临床试验,除非NHP是唯一的相关和敏感物种,该指南再次强调3R (reduce减少,refine优化, replace替代)。
2022年10月31日,FDA 采用CDER专家访谈的形式,请FDA/CDER/OND药理学和毒理学副主任Timothy McGovern博士讨论了该指南对患者、药物研发行业和公众的意义。
2022年11月01日,FDA再次发布了S1B药物致癌性试验研究S1B(R1)附录《S1B(R1) Addendum to S1B Testing for Carcinogenicity of Pharmaceuticals》。
以下是鼎泰团队对Tim 博士的访谈实录概括。
Q
A
问:为什么FDA和ICH认为需要这个指南?
答:在FDA CDER,我们一直在寻找如何最大程度的改善安全性评估和减少对动物试验的,而同时保证最终使用这些药物的患者的安全。基于这一点,我们与ICH以及关心药物研发安全的个人和团体合作,对药品致癌性试验的安全指南做了修订。
提供一些背景信息,早在1995年和1997年就发表了 ICH S1指南(致癌性研究安全指南),阐述了评估新药致癌风险的策略和重要性。策略要求在一种啮齿动物中进行一项长期试验和第二个另一种啮齿动物短期或长期补充试验。该策略通常指为期两年的大鼠致癌试验和为期两年的小鼠或为期六个月的转基因(或基因改造)小鼠的致癌试验。
自1997年ICH S1指南定稿以来,寻求纳入新的科学方法,改进现行模式的努力一直都没有停止过。我们一直致力于减少药物开发研究中所需要使用的动物。
问:您是如何制定指南的?证据权重概念是如何被引入的?
答:对致癌性机制更深入的了解,包括几项回顾性分析的发表,提示在某些情况下,为期两年的大鼠致癌试验可能不会增加对人类致癌风险评估的价值。我们需要把患者安全、加快药物开发和动物福利的综合考量,是我们修改S1B指南的初衷。在ICH框架下,FDA参加了一个专家工作组,通过回顾性数据分析和一项前瞻性研究,以确定现行的两年大鼠致癌性研究的替代方案是否仍然可以充分评估药物的致癌风险。我们最终确定,并在指南中描述,利用“证据权重”方法可以判断在某些情况下无需使用两年大鼠致癌试验,仍可以对药物致癌风险给与充分评估。
从科学术语字面上讲,证据权重是指调查人员对所有相关证据进行系统评估来得出答案或者判断。好比是一个天平,把每一项证据都放在天平称一称,最终(所有证据汇总)提示一个临界点,帮助我们得出(正确)结论。
我们的证据权重评估的数据中包括有关药物的作用机制,毒理学特征,遗传毒性的可能性,免疫调节(或变化)的证据,以及激素失调的证据等。该指南更详细地介绍了上述因素和可推荐考虑的其他因素。
问:究竟如何在评估致癌风险方面应用证据权重方法呢?
答:概括来说,证据权重方法有助于我们获得以下三个结论之一:
对证(数)据权重分析能够提示产品可能存在致癌风险。在我们已经确定看预见高致癌风险情况下,两年的大鼠致癌试验就失去了意义。
证据权重分析能够表明致癌风险不大。在已经确定了低致癌风险情况下,为期两年的大鼠致癌试验也是不必要的。
如果对已有数据的综合权重分析的结果表明致癌风险是模棱两可的,为期两年的大鼠致癌试验是有益的,它可以帮助确定风险的程度。
问:您认为该指南将如何帮助减少用动物进行试验?
答:指南推荐的证据权重法刚刚开始,可减少动物的数目还难以给出具体的数字。但指南专家小组的前瞻性研究估计,综合证据权重法应能将致癌性研究中使用大鼠的数目减少约25%左右。我还想强调,指南继续推荐小鼠致癌性研究仍然是致癌性评估的组成部分。药物研发者有责任开展小鼠致癌风险的研究。因此,指南是改进致癌性这一重要安全终点的评估并考虑动物替代方案方面进一步努力的结果。在考虑药物安评的替代方法或者引入其他方法时,必须继续谨慎行事,始终将患者安全放在首位。
问:您还想从指南中强调其他问题吗?
答:当然。该指南还介绍了基于药物暴露量来设计rasH2-Tg小鼠模型中高剂量的方法。这种方法已用于为期两年的致癌试验,但尚未用于六个月的转基因小鼠的致癌试验。指南给出结论是,用高于人的50倍暴露量作为该模型高剂量选择的标准。当然,研发者还要遵从S1指南中的其他标准。
03
结语
鼎泰集团作为专病领域研发赋能新型CRO,致力于从疾病机理、疾病模型到非临床研究,再到临床转化的深度赋能,多年以来服务于众多企业/科研院所,积累了丰富的研究经验。药物研发,道阻且长,综合国内CDE、FDA的专家意见,关于药物致癌性试验的开展,应充分评估其必要性、重要性、科学性、资源可及性,依托于上述决策树与监管机构保持沟通。
参考资料:
[1] (CDER访谈英文原文来自美国FDA网站https://www.fda.gov/drugs/news-events-human-drugs/cder-conversation-fdas-final-guidance-carcinogenicity-testing-pharmaceuticals)
[2] S1B(R1)药物致癌性S1B试验的修订指南原文美国FDA网站FDA Guidance for Industry-S1B(R1) Addendum to S1B Testing for Carcinogenicity of Pharmaceuticals; International Council for Harmonization accessed on November 19, 2022 at https://www.fda.gov/media/152777/download)
[3] FDA Guidance for Industry-Nonclinical Considerations for Mitigating Nonhuman Primate Supply Constraints Arising from the COVID-19 Pandemic (FEBRUARY 2022, accessed on November 20, 2022 at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/nonclinical-considerations-mitigating-nonhuman-primate-supply-constraints-arising-covid-19-pandemic )
[4] FDA: Advancing Alternative Methods at FDA (accessed on November 20, 2022 at https://www.fda.gov/science-research/about-science-research-fda/advancing-alternative-methods-fda)
[5] Timothy McGovern,WEIGHT OF EVIDENCE APPROACHES FOR EVALUATING CARCINOGENESIS IN DRUG DEVELOPMENT (APRIL 29, 2019) (accessed on November 20, 2022 at https://ntp.niehs.nih.gov/events/webinars-workshops/2019/coc/presentations/10mcgovern_coc_508.pdf)







 法规政策
法规政策 项目管理
项目管理


 Global
Global



 苏公网安备32011202001081
苏公网安备32011202001081