



前两期文章中,鼎泰集团先后对第1款和第2款已上市 siRNA 药物 —— Patisiran(Onpattro®,点击查看原文) 和 Givosiran(Givlaari®,点击查看原文) 的上市历程及临床开发路径进行了复盘分析,并尝试对与临床转化有关的内容进行了总结,为 siRNA 药物运用临床前研究结果指导临床开发,以及制定有助于产品快速上市的研发和药政策略提供了参考和启发。
作为第3款获批上市的 siRNA 药物,Lumasiran 从 IND 申请到获批上市同样仅用了4年的时间,并被 FDA 批准同时用于治疗成人和儿童原发性高草酸尿症1型(PH1)。这是 FDA 批准的首款治疗罕见代谢紊乱疾病的药物,再次彰显了 siRNA 药物在治疗罕见疾病方面的巨大潜力。
作为一款同时获批用于成人和儿童患者的 siRNA 药物,Lumasiran 的开发路径与 Patisiran 和 Givosiran 相比有哪些独特之处?在本期内容中将为您揭晓。
★ 文章导览 ★
01 | 原发性高草酸尿症1型(PH1) |
02 | Lumasiran 产品信息 |
03 | 临床开发路径 关键词:I/II 期无缝隙设计;2项关键III期 |
04 | 监管互动里程碑 关键词:EOP2;幼龄动物研究;致癌性研究 |
05 | 药代动力学与免疫原性检测 |
06 | 非临床与临床试验相关性分析 |
原发性高草酸尿症1型(PH1)

PH1 发病机制示意图
Lumasiran 产品信息
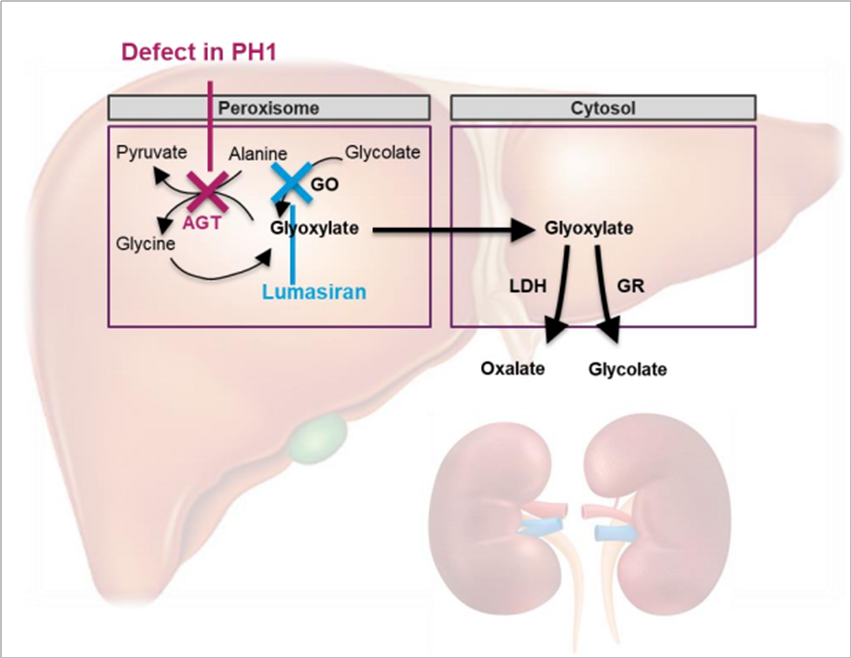
Lumasiran 作用机理示意图
Lumasiran 临床开发路径
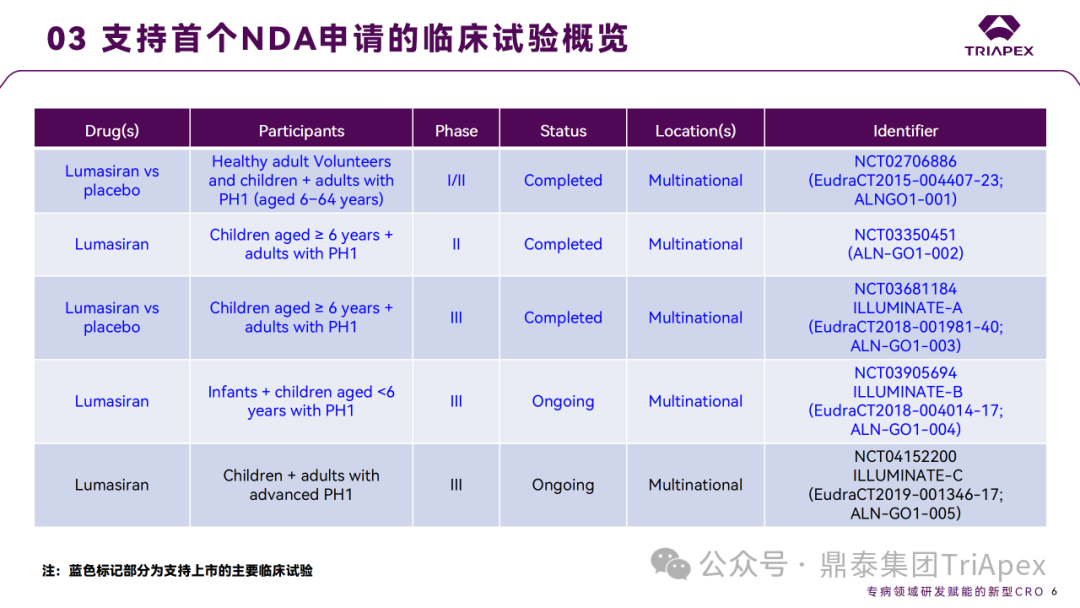
支持首个 NDA 申请的临床试验
◆ I期临床试验(NCT02706886)主要目的是评估 Lumasiran 在健康受试者和 PH1 患者中的安全性和耐受性。
◆ II期扩展研究(NCT03350451)评估 Lumasiran 对 PH1 成人和儿童的长期和安全性和耐受性,研究完成日期为2023年6月。
◆ 2项 Ⅲ 期临床试验 ILLUMINATE-A(NCT03681184)和 ILLUMINATE-B(NCT03905694)分别评估 Lumasiran 治疗6岁及以上 PH1 患者和小于6岁的婴儿和儿童 PH1 患者的长期有效性和安全性,其中 ILLUMINATE-A 完成时间为2024年1月;提交 NDA 时,ILLUMINATE-B 还在进行中,提供了初始6个月的部分数据以支持6岁以下儿童用药,完整试验预计2024年8月完成。
Lumasiran 临床开发路径
1
Ⅰ/Ⅱ 期临床试验(NCT02706886)

Ⅰ/Ⅱ 期临床试验设计


Ⅰ/Ⅱ 期临床试验分组和给药方案
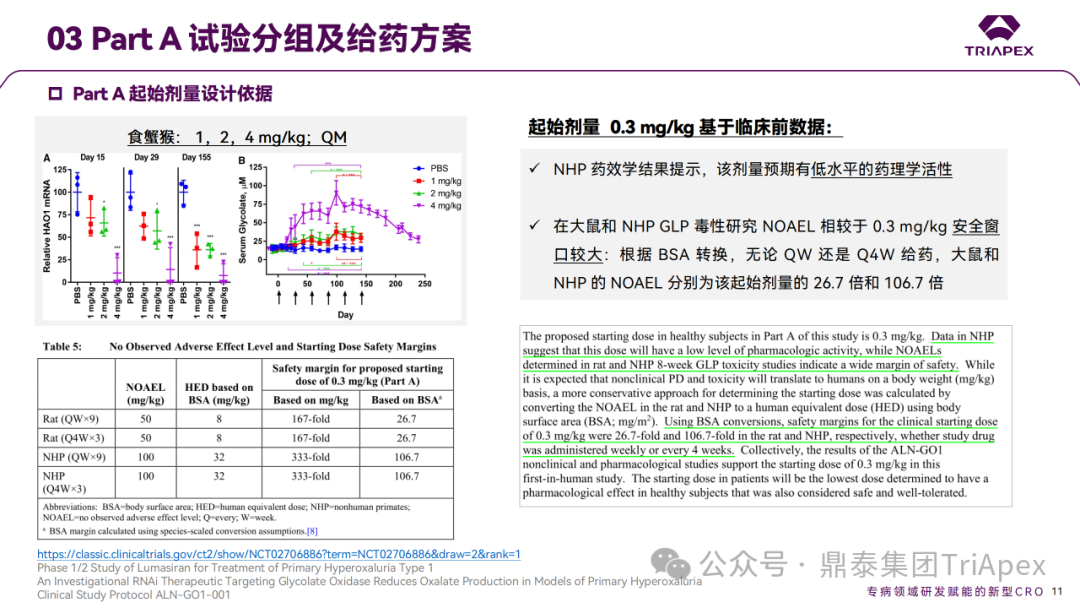
Part A 起始剂量设计依据
Part B起始剂量和给药频率的设计依据:
根据 Part A 数据,1 mg/kg 是健康受试者中具有药理作用且耐受性良好的最低剂量。
给药间隔的设计主要基于临床前数据和 Part A 结果。临床前数据显示,为了维持靶点的有效抑制,需要 QM 的给药频率;Part A 的初步数结果也为 Part B 的 Q3M 给药方案提供了依据(笔者猜测可能基于 Part A 中单次给药后,于 D85 时血浆和尿乙醇酸仍能保持较高水平)。

Part B 起始剂量设计依据
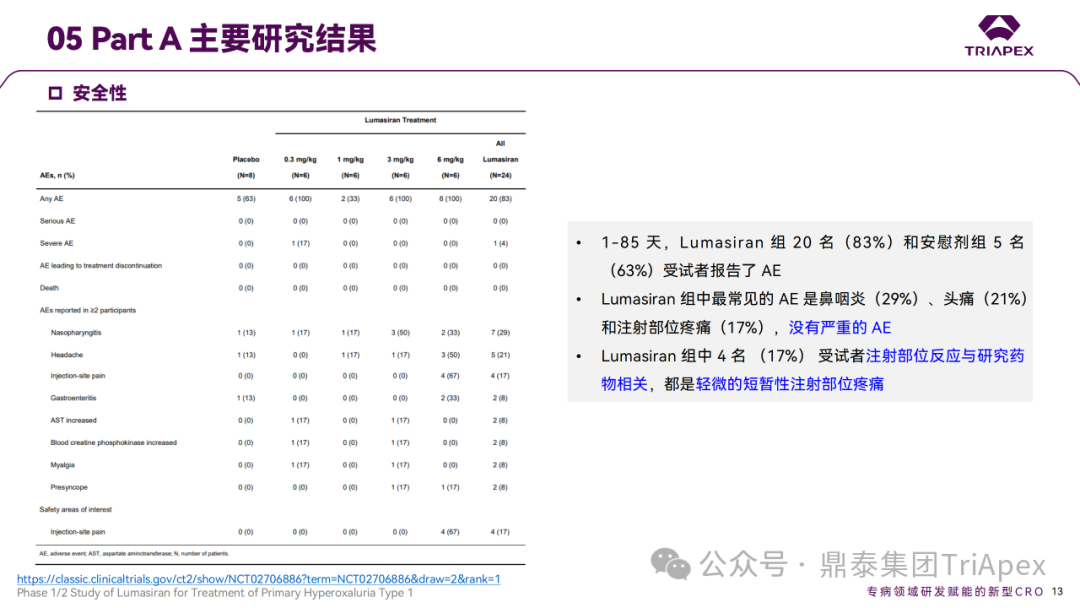
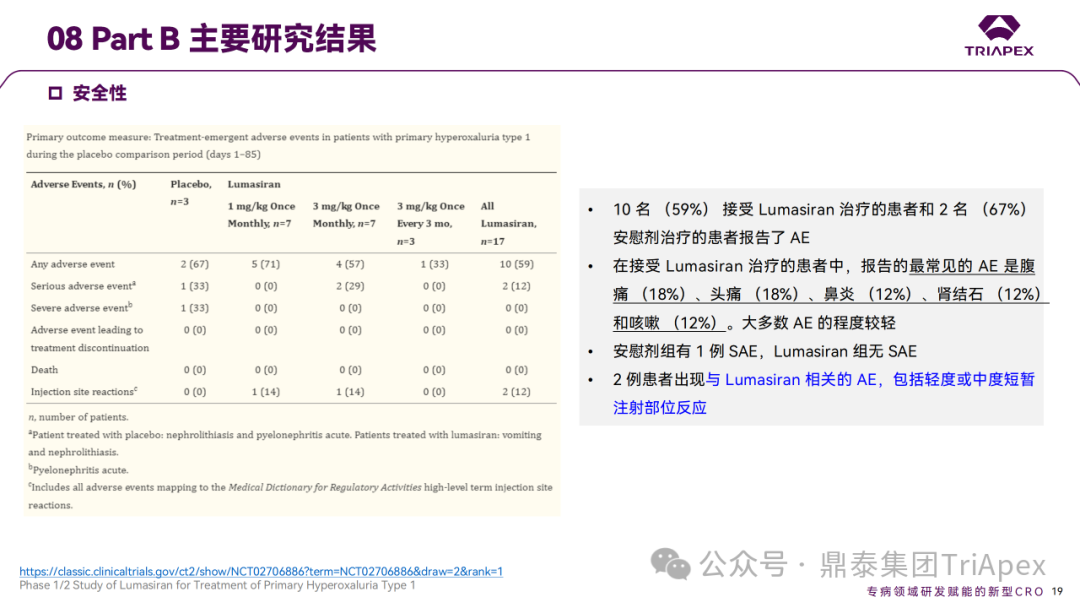
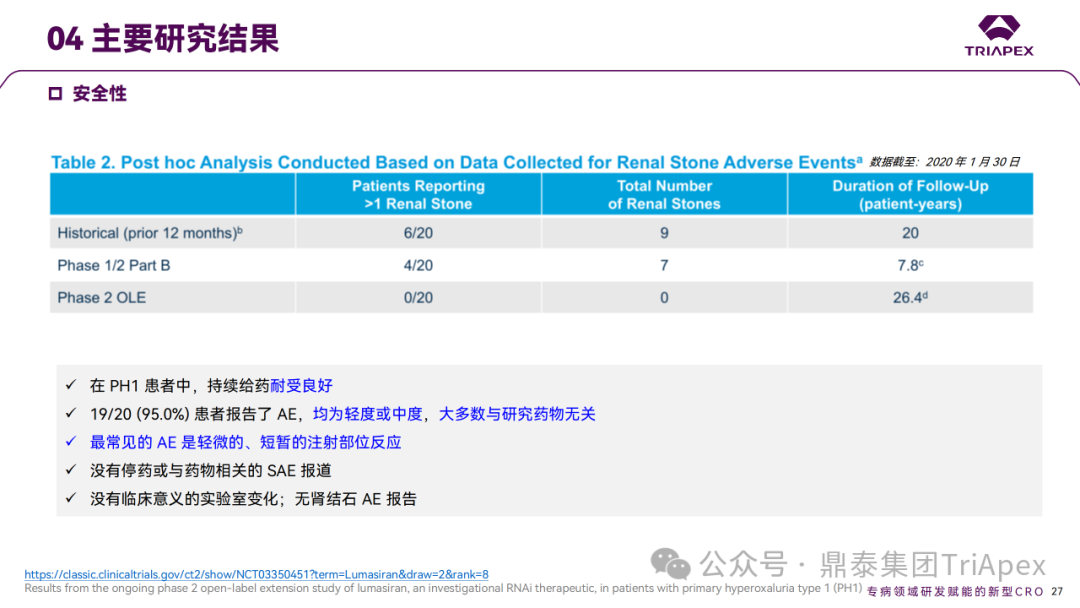
Ⅰ/Ⅱ 期临床试验安全性相关结果
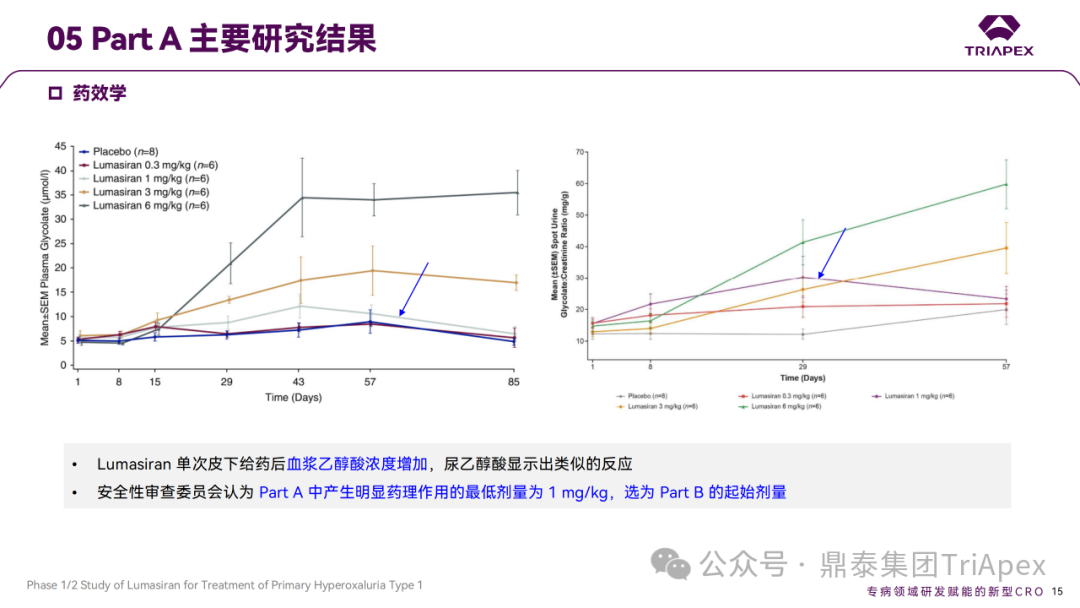
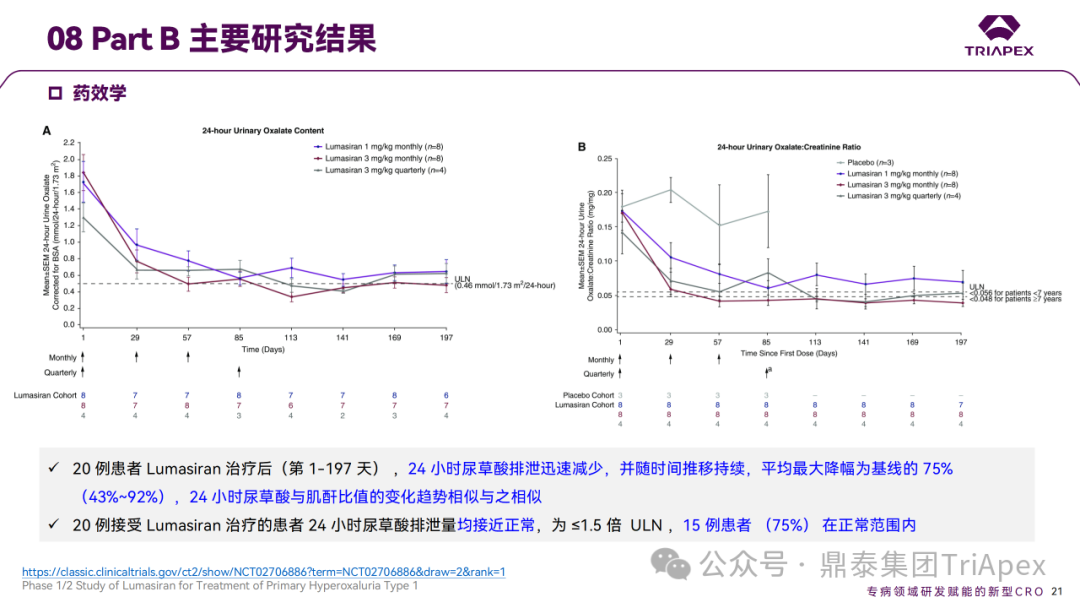
Ⅰ/Ⅱ 期临床试验药效学相关结果
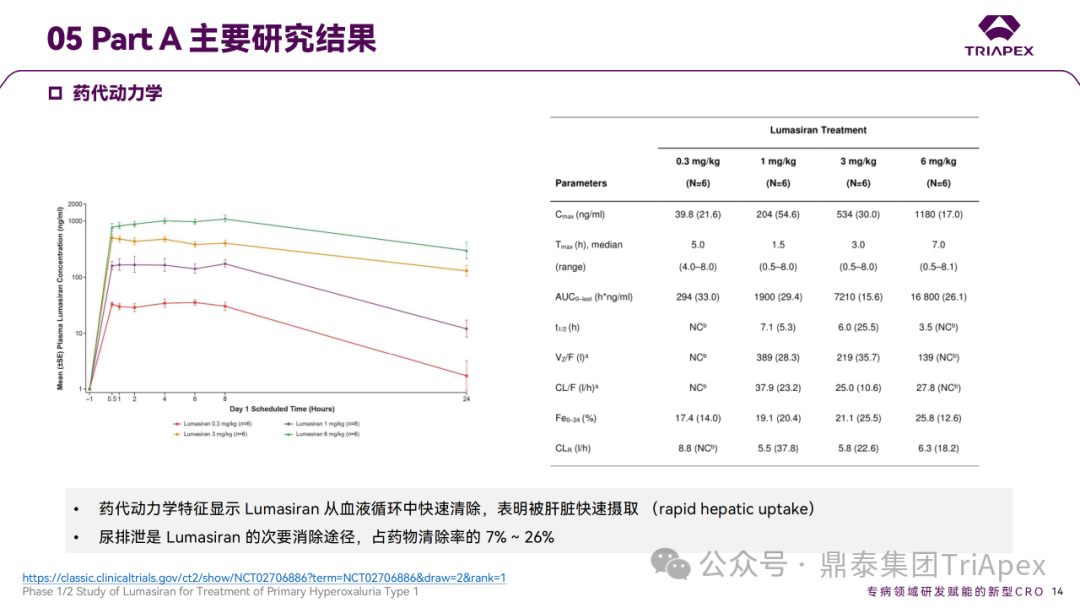
Ⅰ/Ⅱ 期临床试验药代动力学相关结果
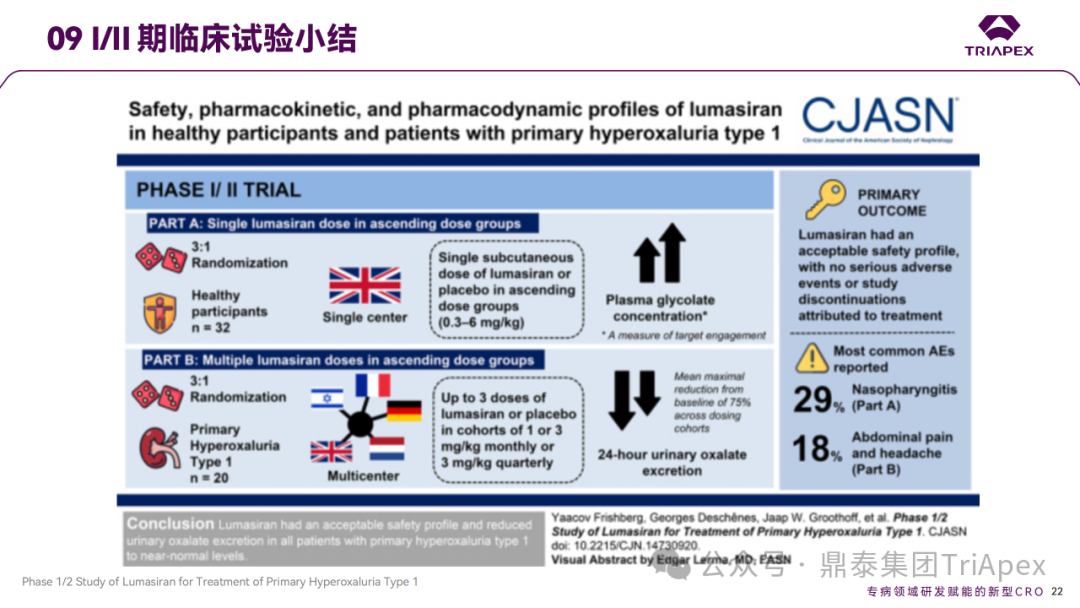
Ⅰ/Ⅱ 期临床试验小结
2
Ⅱ 期临床试验(NCT03350451)


Ⅱ 期临床试验设计
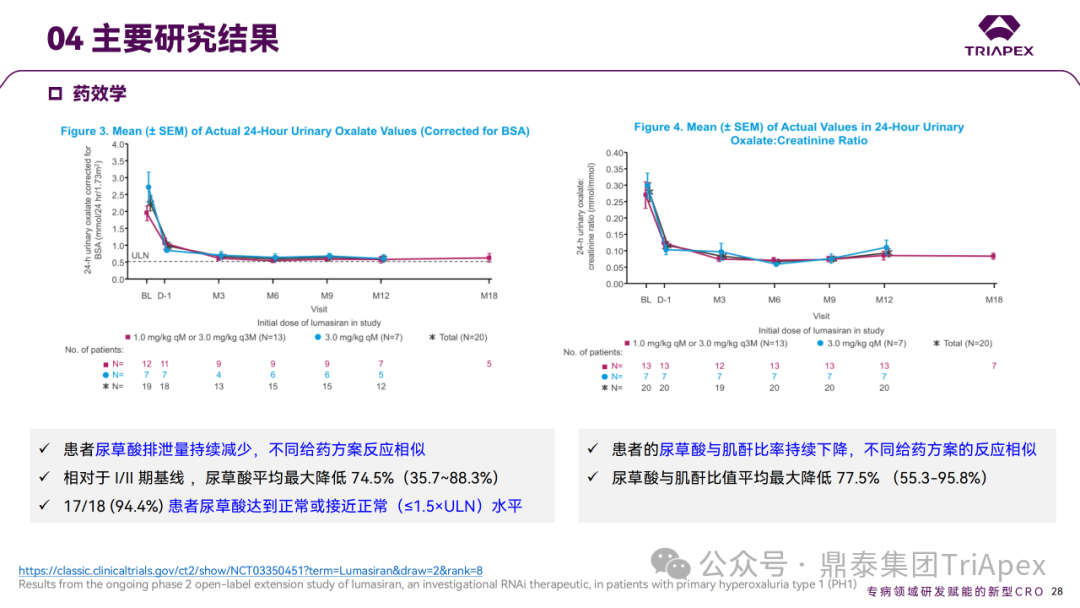
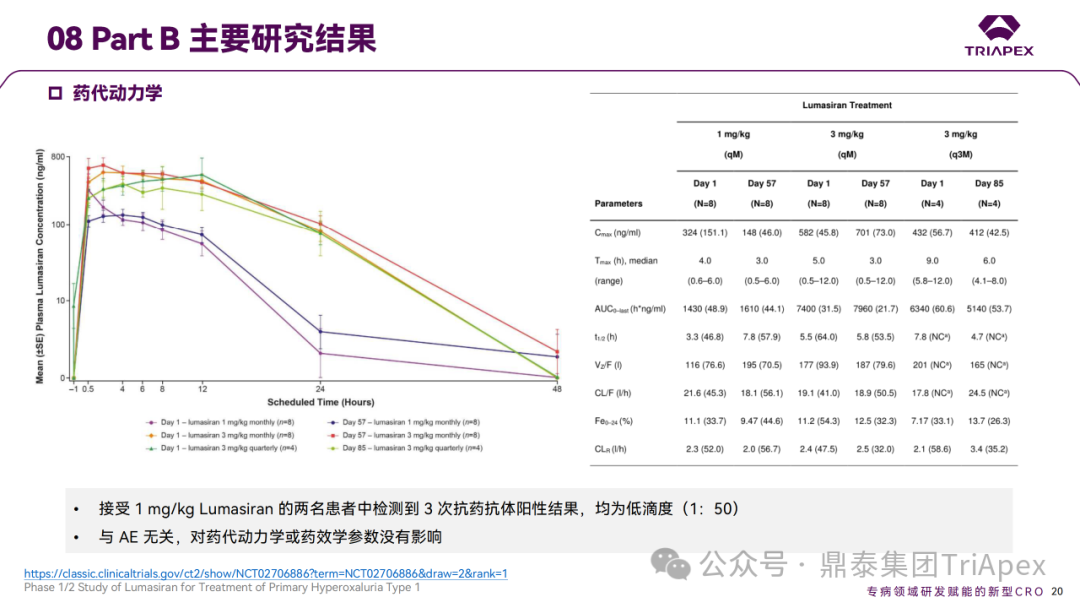
Ⅱ 期临床试验研究结果
3
Ⅲ 期临床试验——
ILLUMINATE-A(NCT03681184)
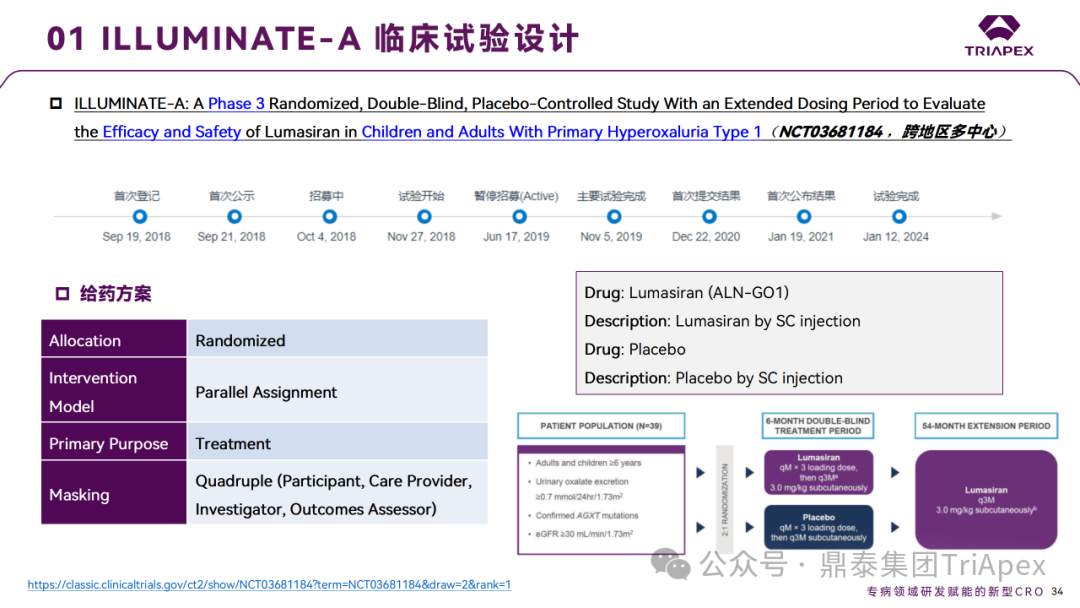
Ⅲ 期临床 ILLUMINATE-A 试验设计
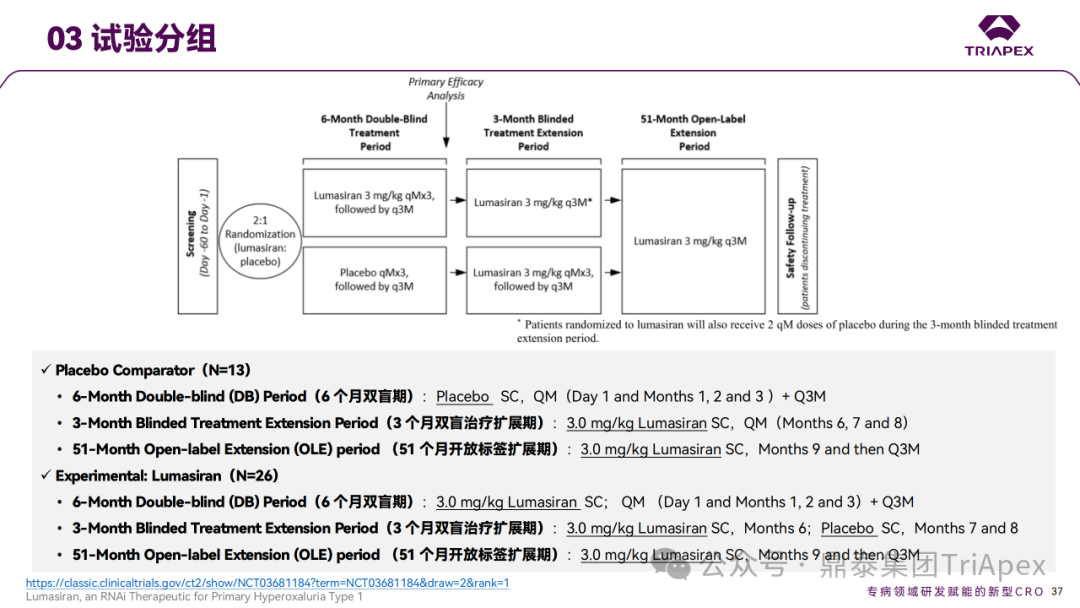
Ⅲ 期临床 ILLUMINATE-A 试验分组
6岁及以上的 PH1 患者III期临床试验剂量和频率设计依据:
剂量设计主要基于研究 ALN-GO1-001 Part A 和 Part B中健康受试者和 PH1 患者的数据以及相关的 PK/PD 模型。在 PH1 患者中,群体 PK/PD 模型显示在剂量范围(1.0~6.0 mg/kg,QM 或 Q3M,SC)草酸呈剂量依赖性减少,随着剂量的增加,预期的附加效应逐渐减弱。
对于给定剂量,PK/PD 模型显示,与 Q3M 给药相比,QM 给药导致尿草酸水平下降更快。以 QM 负荷剂量 + Q3M 维持剂量给药,可以实现 PH1 患者尿草酸快速和持续降低到正常或接近正常水平的目标。

Ⅲ 期临床 ILLUMINATE-A 剂量设计依据
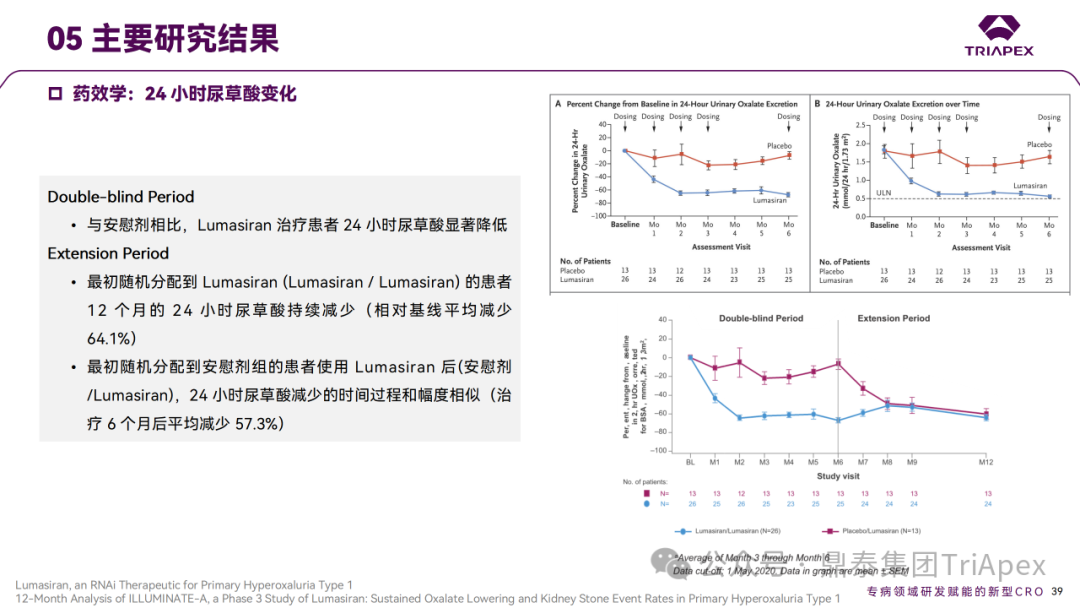

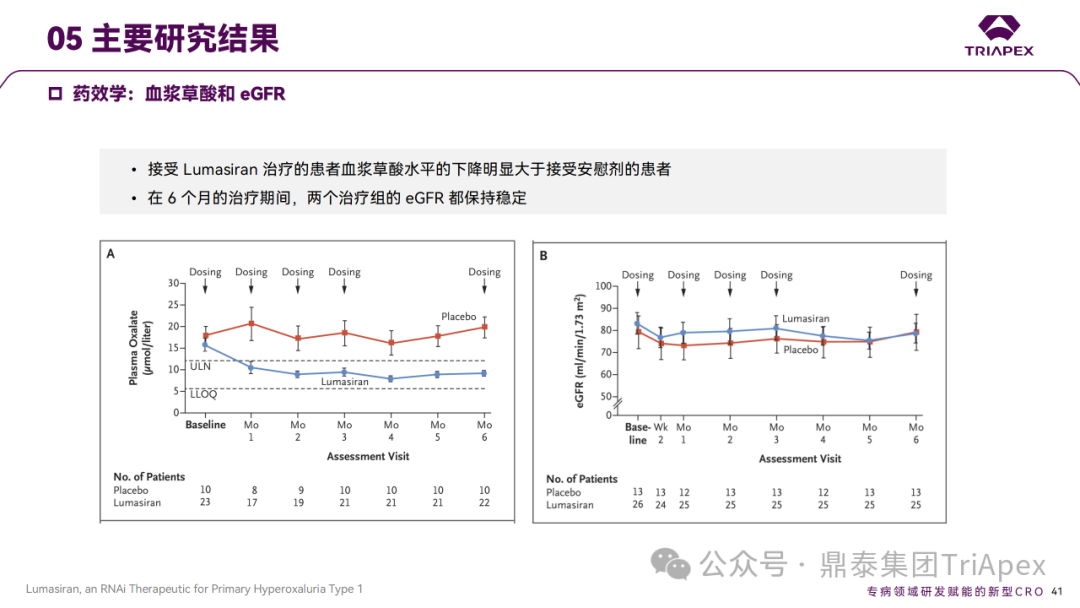

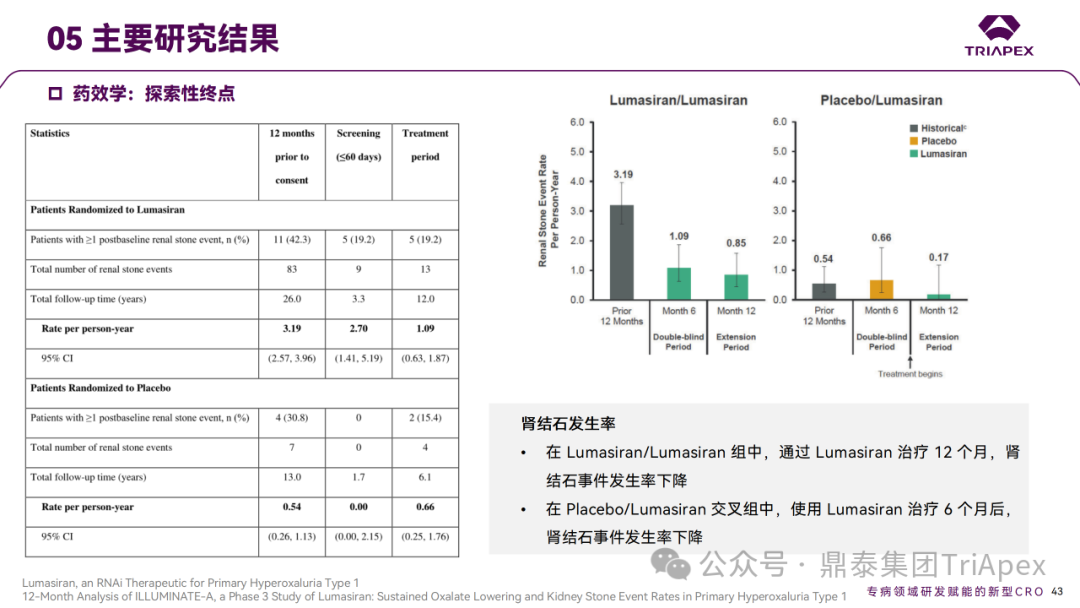
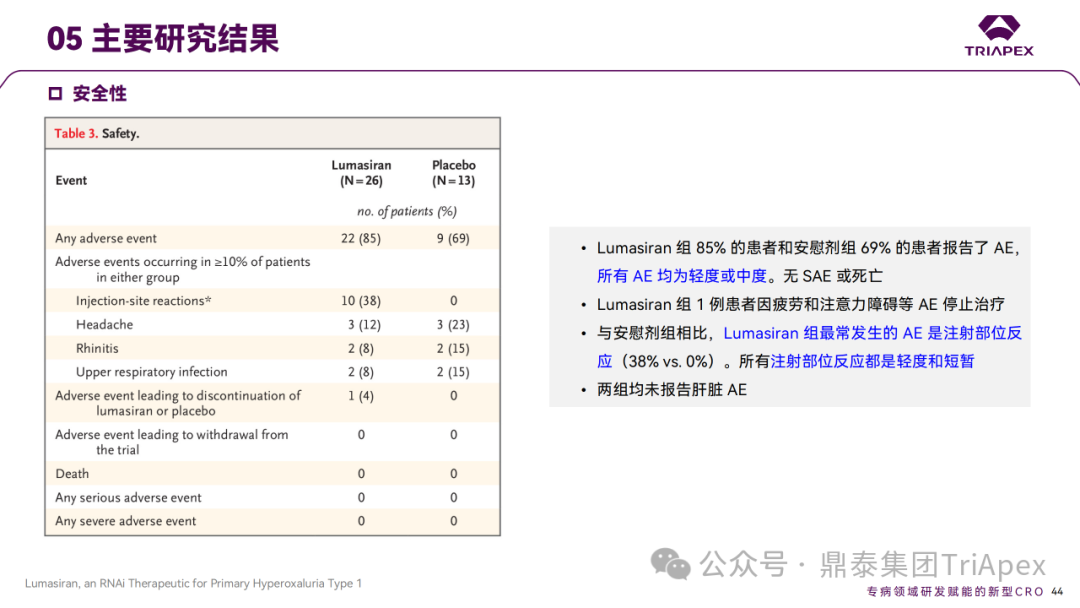
Ⅲ 期临床 ILLUMINATE-A 主要研究结果
4
Ⅲ 期临床试验——
ILLUMINATE-B(NCT03905694)

Ⅲ 期临床 ILLUMINATE-B 试验设计
婴幼儿患者III期临床试验剂量和频率设计依据:
本试验纳入18名0~5岁 PH1 婴幼儿患者,使用临床前数据和 Ⅰ/Ⅱ期试验 PH1 患者的临床数据以支持 PK/PD 建模和剂量选择;
考虑到儿童生长速度快、肝脏相对重量较大、药物清除速度快,需要更高的剂量才能达到类似的全身浓度和靶点抑制。
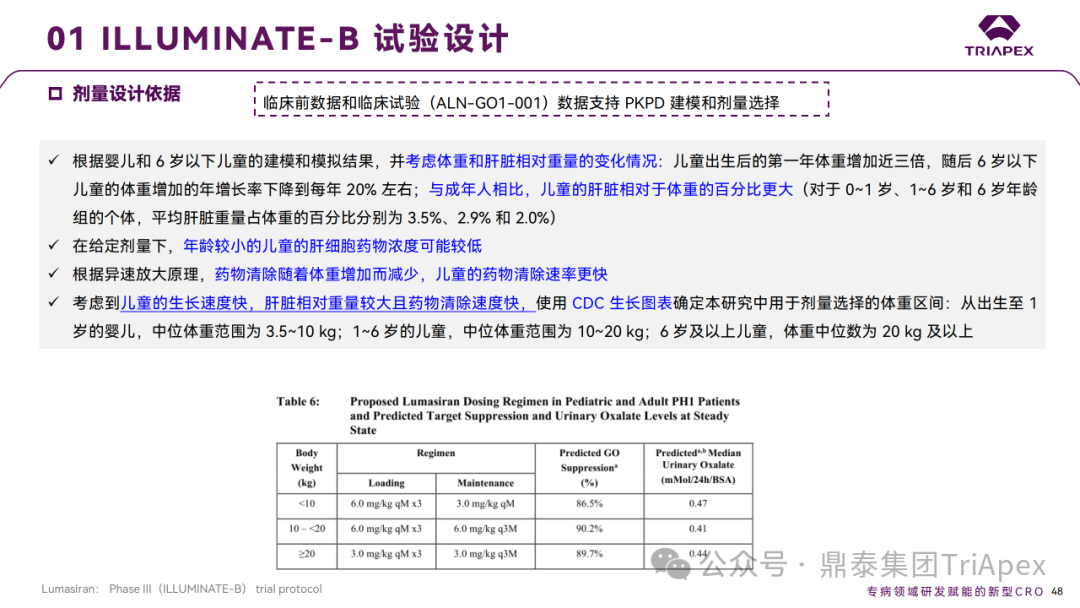
Ⅲ 期临床 ILLUMINATE-B 剂量设计依据
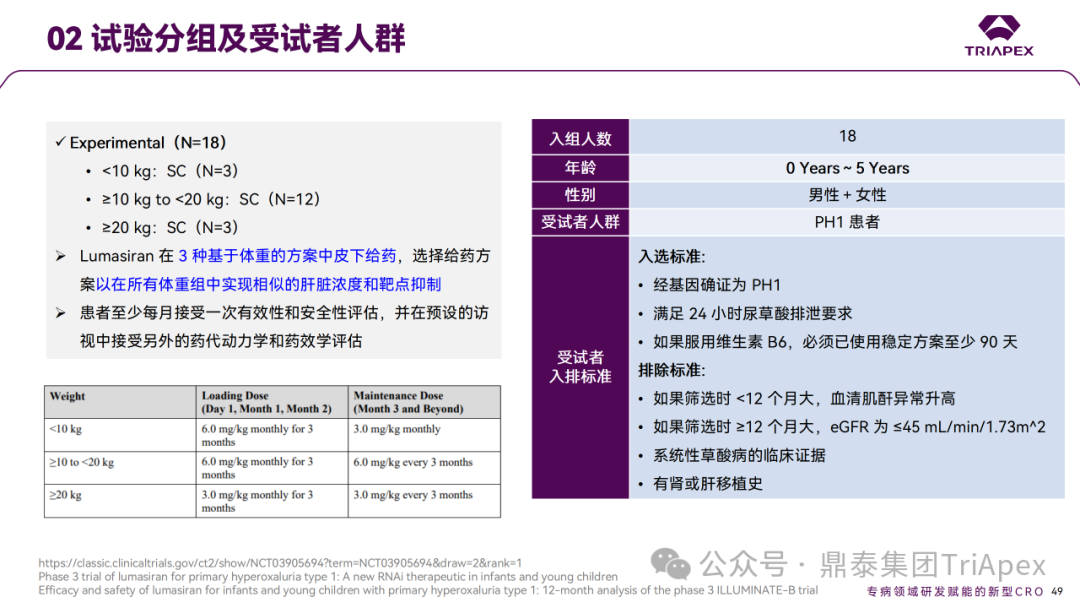
Ⅲ 期临床 ILLUMINATE-B 试验分组及受试者人群
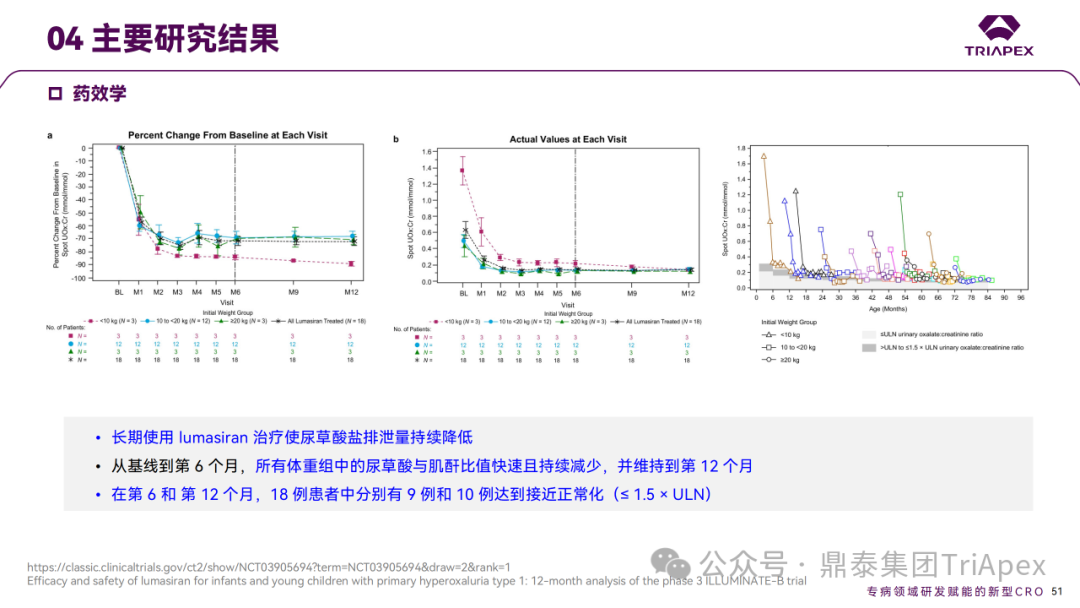
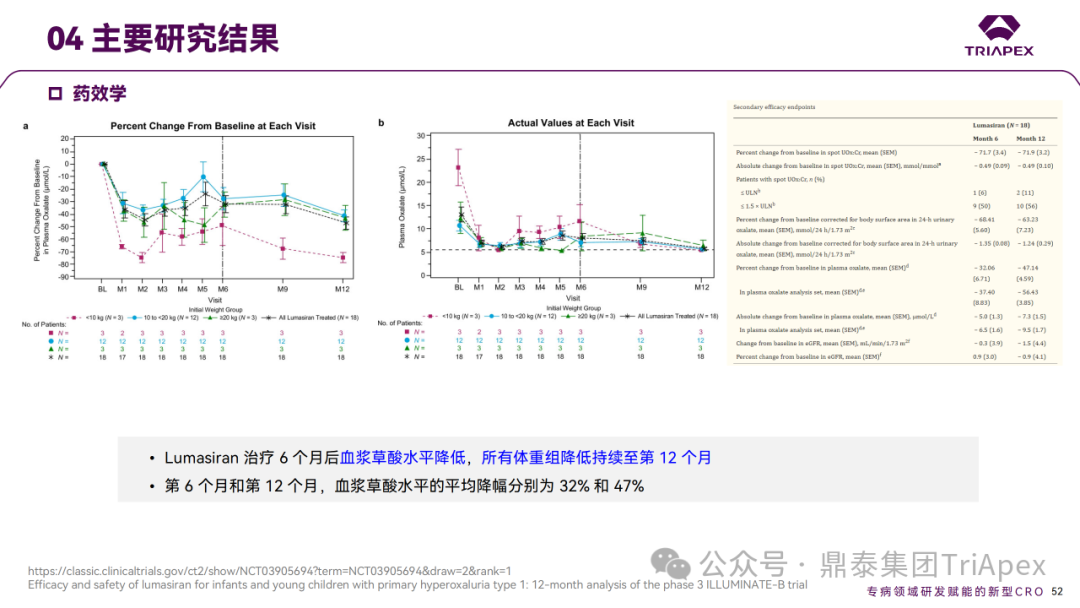
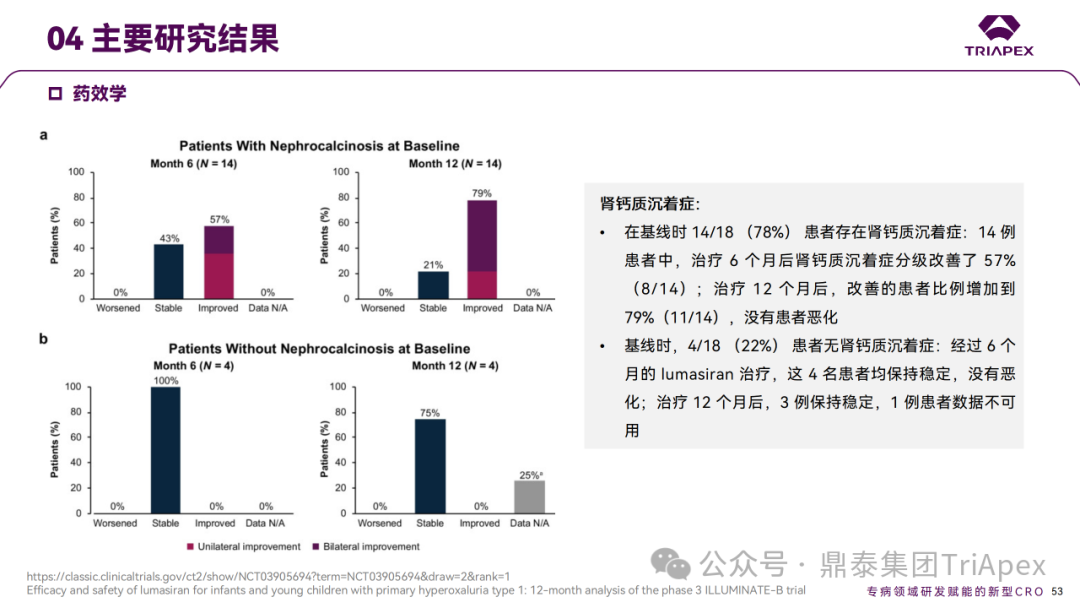
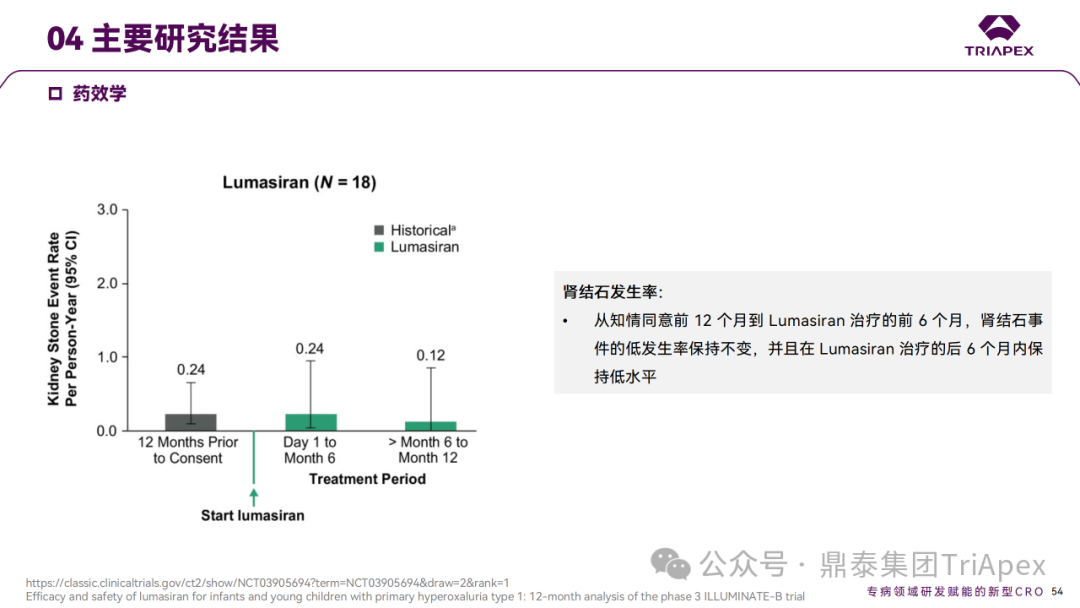
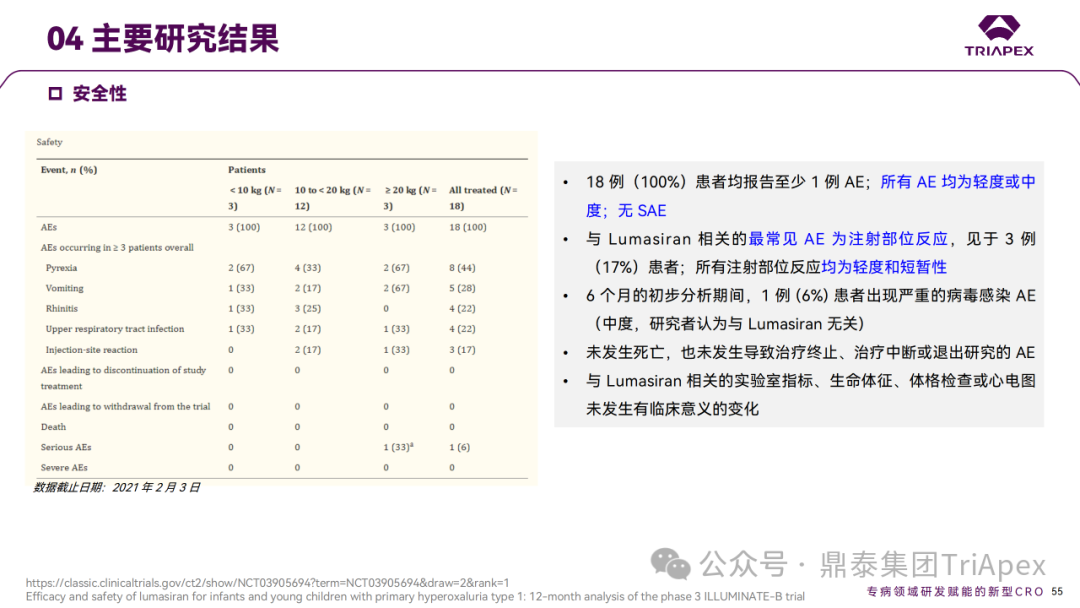
Ⅲ 期临床 ILLUMINATE-B 主要研究结果
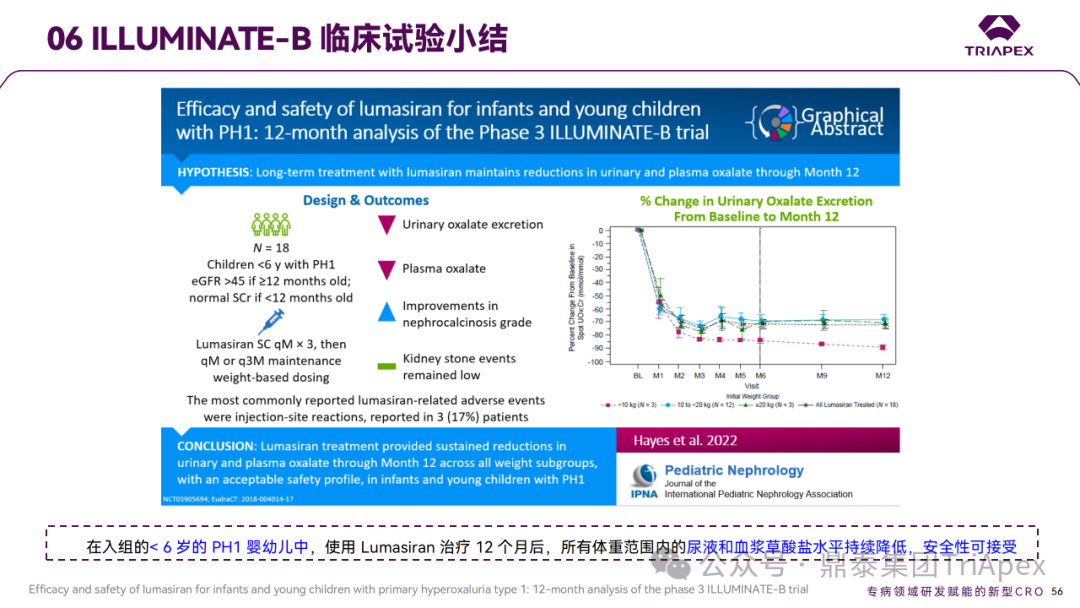
Ⅲ 期临试验床 ILLUMINATE-B 小结
结语
参考资料:
[1] Phase 1/2 Study of Lumasiran for Treatment of Primary Hyperoxaluria Type 1
[2] Lumasiran, an RNAi Therapeutic for Primary Hyperoxaluria Type 1
[3] Results from the ongoing phase 2 open-label extension study of lumasiran, an investigational RNAi therapeutic, in patients with primary hyperoxaluria type 1 (PH1)
[4] 12-Month Analysis of ILLUMINATE-A, a Phase 3 Study of Lumasiran: Sustained Oxalate Lowering and Kidney Stone Event Rates in Primary Hyperoxaluria Type 1
[5] Phase 3 trial of lumasiran for primary hyperoxaluria type 1: A new RNAi therapeutic in infants and young children
[6] Efficacy and safety of lumasiran for infants and young children with primary hyperoxaluria type 1: 12-month analysis of the phase 3 ILLUMINATE-B trial







 法规政策
法规政策 项目管理
项目管理


 Global
Global



 苏公网安备32011202001081
苏公网安备32011202001081