


“寡核苷酸药物(Oligonucleotide Therapeutics,ONTs)是一种新兴的治疗手段,包括各种人工合成的 RNA 或 RNA/DNA 杂交分子,通过结合靶 RNA 序列而改变 RNA 表达和/或下游蛋白表达。ONTs 在作用机制、结构、化学修饰、分子量、序列、给药策略和偶联物等方面不同于常规化药,且不同分子类型之间也存在差异,这决定了 ONTs 在人体内和不同人群之间可能具有独特的行为学。”
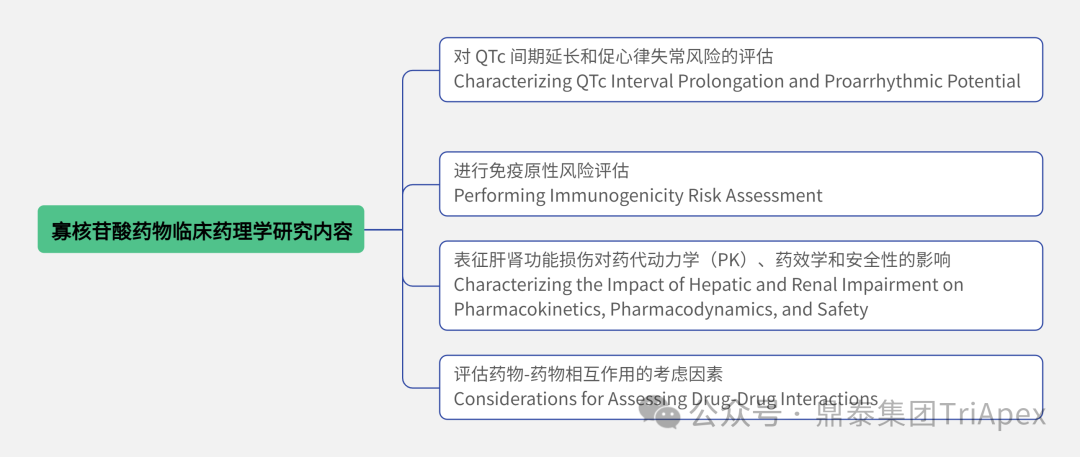
经过 2 年的征求意见,FDA CDER 最终于 2024 年 6 月 14 日发布了 Guidance for Industry《Clinical Pharmacology Considerations for the Development of Oligonucleotide Therapeutics》。该指导原则为 ONTs 开发过程中与临床药理学研究相关的关键问题给出了 4 个方面的建议,并阐述了何时进行上述研究以及适合开展的试验类型。 对 QTc 间期延长和促心律失常风险的评估 Characterizing QTc Interval Prolongation and Proarrhythmic Potential 进行免疫原性风险评估 Performing Immunogenicity Risk Assessment 表征肝肾功能损伤对药代动力学(PK)、药效学和安全性的影响 Characterizing the Impact of Hepatic and Renal Impairment on Pharmacokinetics, Pharmacodynamics, and Safety 评估药物-药物相互作用的考虑因素 Considerations for Assessing Drug-Drug Interactions
I.适用范围 该指南适用于通过 Watson-Crick 碱基配对靶向 RNA 的 ONTs;直接调节蛋白(如 Aptamer)、免疫激活剂(如 TLR9 激动剂)或 RNA/DNA 编辑(如 CRISPR)等作用机制的 ONTs 不在本指南范围内。随着技术的进步,对于特定的化学修饰、结构或给药策略等也不断完善,指南建议就相关问题及时与 FDA 进行沟通。 II. 临床药理学研究内容 一般而言,ONTs 在体循环中会被快速清除,但在组织中的分布和累积水平较高并具有较长的药效学半衰期,导致持续的药效学反应。因此,在确定临床药理学研究的内容时,需考虑以下因素: 在药物早期开发阶段,考察 ONTs 经单次给药和多次给药后的 PK 特征。 考虑到 ONTs 的全身 PK 数据可能无法反映其在靶组织中的分布、药效学或有效性、安全性,FDA 推荐评估适当的药效学生物标志物。如在多次给药研究(multiple-dose studies)中,尽可能评估合适的药效学生物标志物(如靶 mRNA、靶蛋白或反映对靶蛋白进行调控后的下游生物标志物)或其他指标。该研究在药效学变化与全身 PK 变化不一致的情况下尤为重要。药效学终点的选择应与 FDA 进行讨论,特别是当药效学终点可能无法直接反映靶组织中的中靶点抑制情况时(如 CNS 靶点的脑脊液、肝脏靶点的血浆)。 与小分子药物或生物制品相比,ONTs 在化学合成、结构、作用位点、PK 和药效学方面具有独特之处。在进行上述研究时,应考虑 ONTs 的化学修饰、靶点、蛋白结合率和给药途径等因素,因为上述因素主要决定了其在肝肾和其他组织中的分布以及全身或局部的暴露水平。 使用合适的生物分析方法测定 ONTs 原形药和相关代谢物(如适用)的浓度。 A. 对 QTc 间期延长和促心律失常风险的评估 尽管 FDA 已审评过的少数专门的 QT 研究显示,ONTs 对 QTc 间期的影响较小,但由于 ONTs 种类繁多,现有的临床经验尚不足以对特定类型 ONTs(如基于新的化学结构或递送策略)的心律失常风险做出总体结论。因此,创新 ONTs 在上市前应充分评估其对 QT/QTc 间期的影响,具体可按照 FDA 指南或 ICH E14 进行。评估计划应充分论证并与 FDA 讨论,评估的时间和范围取决于 ONTs 的获益风险状况。
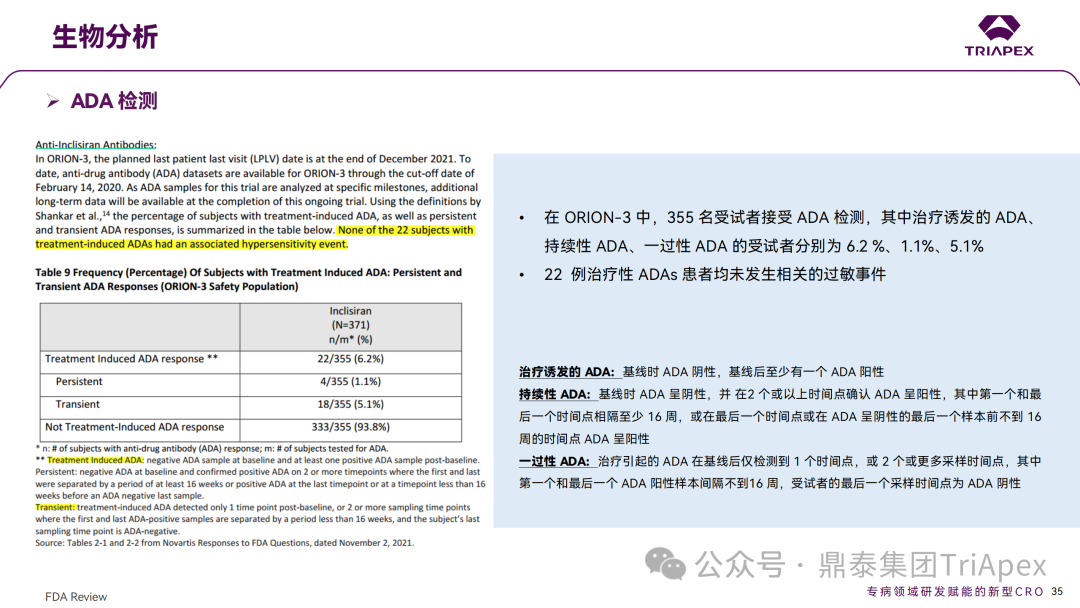
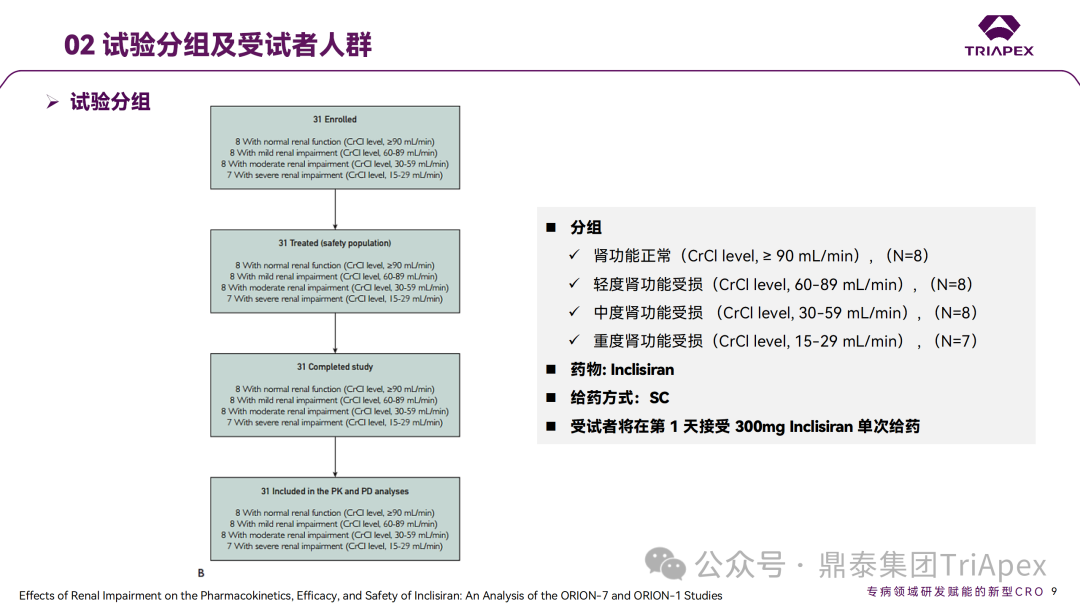
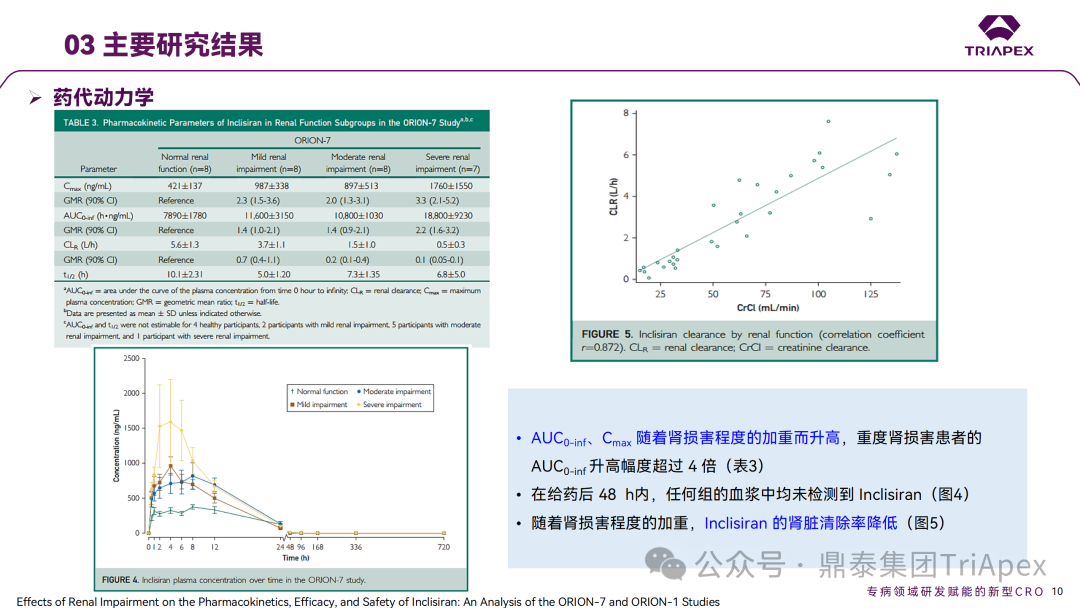
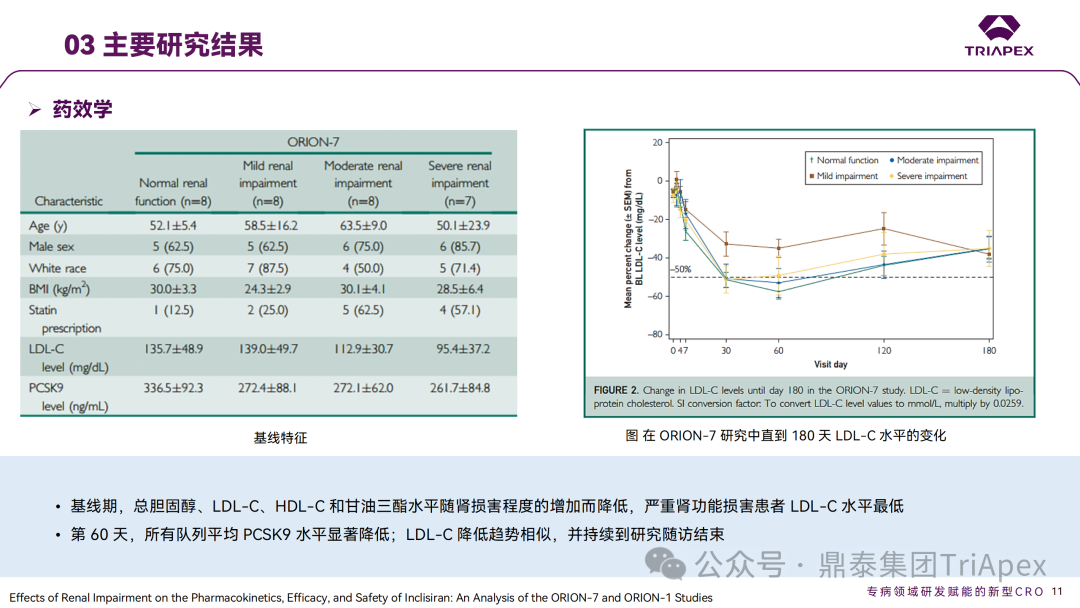
◆ 案例分析: 案例1:Vutrisiran 的 ALN-TTRSC02-001 试验中,评估了对 QT 间期延长的影响。这是一项 1 期、随机、单次递增剂量研究(25mg~300mg),旨在评估健康受试者皮下注射 Vutrisiran 的安全性、耐受性、PK 和药效学,本试验中的最高剂量足以涵盖临床试验最高暴露情况。结果显示,在 9 倍的临床推荐剂量下(25mg,Q3M),Vutrisiran 未造成显著的 QTc 间期延长。该结果也与非临床安全药理学研究结果相吻合。 案例2:在一项专门的 TQT 研究(ORION-12)中,评估了 Inclisiran 对 QTc 间期延长的影响。该研究采用了标准设计,给药剂量为 900mg(超过治疗剂量),以覆盖最差情况下的暴露场景(如肾功能不全)。研究结果显示,在给药 4h 后,Inclisiran 组相比对照组的 QTcF 间期变化(ΔΔQTcF)为 2.9ms,90% 置信区间为 0.5~5.2ms。该结果表明,即使在高于治疗剂量下,Inclisiran 也不会导致有临床意义的 QTc 间期延长。FDA 的 QT-IRT 审评小组于 2020 年 5 月 13 日对该研究进行了详细分析和审查,进一步支持了 Inclisiran 在治疗剂量范围内不太可能产生显著的 QTc 延长效应的结论。这一发现对 Inclisiran 的心脏安全性评估具有重要意义。 摘自 Inclisiran 临床药理学审评报告 B. 进行免疫原性风险评估 ONTs 可能诱导对载体、骨架、序列或新抗原表位的免疫反应。由于该领域发展迅速,新的化学修饰和递送系统会对特定产品的免疫原性风险和临床免疫原性评估方法带来影响。FDA 建议采用基于风险的免疫原性评估方法,并按照 FDA guidance《Immunogenicity Assessment for Therapeutic Protein Products(2014)》制定免疫原性风险评估计划,需要考虑的因素包括: 产品因素:碱基序列、碱基修饰、骨架修饰、单链或双链、纯度、修饰后的核苷酸、二级和三级结构、载体组分(如 PEG 化的 LNP)和偶联物(如肽或抗体)等 药理学特性:作用机制、细胞/组织靶点、表达谱、给药途径、给药方案(慢性还是急性) 患者特征:给药群体的免疫激活状态(如自身免疫或炎症性疾病)、可能影响 ADA 发生率或临床结局的合并用药(如化疗药等免疫抑制剂) 临床免疫原性评估通常包括多层级(multi-tiered)的 ADA 检测,可能需要针对 ONTs 的不同组分,如载体(PET 化的 LNP)、偶联的配体靶向蛋白(如 Fab 片段)开发多种检测方法。对于产生修饰蛋白的 ONTs,建议考虑检测针对该蛋白的 ADA 的产生情况。 此外,在适当情况下有必要评估对非预期的天然免疫的激活。有关评估天然免疫激活作用的更多考虑因素,可参考 FDA Guidance《Immunogenicity Assessment for Therapeutic Protein Products(2014)》。在本指南中列出了需要进行先天性免疫激活检测的几种情形: ONTs 诱导细胞因子释放 存在已知在人体内具有免疫刺激活性的序列,如 GU、CpG 或 5'-P 存在 2'-脱氧、2'-OH 或未甲基化 C 的天然核苷 临床免疫原性样本采集应与 PK 和药效学采样点匹配,以评估 ADA 对 PK、药效以及任何免疫介导的不良反应的影响。与蛋白类药物的 ADA 检测类似,需要评估血清中 ONTs 对于检测的影响。根据免疫原性风险评估的结果,在早期开发阶段(如I期临床/首次人体试验),可以考虑储存样本,以备后续发现 PK、药效学或免疫介导不良反应异常时进行检测。申请人应与 FDA 讨论免疫原性风险评估结果,以及如何为特定产品设计临床免疫原性评估方案。 在某些情况下,FDA 还可能建议评估 ONT 序列特异性抗体和/或生物活性(如中和、增强作用)。具体建议会视临床风险等级而定,如 ONT 序列、新型结构或修饰等,可根据具体情况与 FDA 进行讨论。 ◆ 案例分析: 在 Inclisiran 的非临床研究中,采用 ELISA 法分别评估了大鼠和猴重复给药毒性试验中 ADA 产生情况。结果显示,ADA 发生率和滴度均较低,ADA 的产生与剂量无关,对 PD 结果未见影响。此外,Inclisiran 不会刺激小鼠或猴体内促炎细胞因子的释放,未造成猴补体的激活。 在 EOP2 会议中,申请人主张当前的临床研究中暂未在受试者中检测到 ADA。FDA 建议如果在临床中检测到 ADA,可能需要额外的检测(如确定 ADA 是否与内源性物质有交叉反应)来充分评估产品的安全性。由于在目前临床研究中没有检测到 ADA,FDA 没有要求且认为不需要开发这种检测方法(申请人认为由于 RNA 的不稳定性,很难开发出这样的检测方法;在观察到 ADA 的情况下检测免疫复合物已足够)。FDA 建议在第 30 天新增 ADA 的检测,以评估早期 IgG 反应。此外,FDA 还建议申请人在关键临床试验期间收集样品用于 ADA 检测。 在随后的一项 Ⅱ 期(ORION-3)试验中,对 355 名受试者进行了检测,发现治疗诱发的 ADA(Treatment Induced ADA)、持续性(Persistent)ADA、一过性(Transient)ADA 的受试者比例分别 6.2%、1.1%、5.1%,上述患者均未发生相关的过敏事件。在 3 项 Ⅲ 期(ORION-9、ORION-10 和 ORION-11)临床试验中,对 1830 名受试者进行了 ADA 检测,研究结果发现治疗诱导的 ADA 反应发生率为 4.9%,且滴度普遍较低;一过性 ADA、持续性 ADA 的发生率分别为 2.2%、1.7%。此外,申请人在 pre-NDA 会议中指出,ADA 的存在与 AE、LDL-C 和 PSCK9 水平的变化之间没有关联。 详细内容可参考往期文章(点击链接:他山之石-4丨siRNA 药物 Inclisiran 上市历程及临床开发路径总结) 鼎泰集团内部总结 C. 表征肝肾功能损伤对 PK、药效学和安全性的影响 申请人应根据非临床和I期临床试验数据确定肝脏和肾脏在 ONTs 分布、清除和药效方面的作用,从而确定评估肝肾功能损伤影响的方法。 早期 PK、药效学表征以及安全性和耐受性信息,可用于指导后期试验中纳入不同程度肝肾功能损伤受试者。此外,对于肝肾功能损伤受试者,还需考虑药物靶点及偶联物靶点(如用于靶向肝脏的受体)的表达和转换速率(turnover)。 如果 ONTs 不经肾脏清除或不靶向肝脏,后期临床试验通常应纳入肝肾功能正常和不同损伤程度的受试者,以积累在该人群中的安全性和有效性数据。给药剂量应基于非临床和早期临床经验,如需排除肝肾功能损伤受试者,申请人应提供合理依据。更细节的内容,建议参考 FDA Guidance《Enhancing the Diversity of Clinical Trial Populations-Eligibility Criteria,Enrollment Practices,and Trial Designs(2020)》 如果药物主要经肾脏排泄(≥30%),建议评估重度肾功能损伤对 PK、药效动力学和安全性的影响。可考虑采用简化设计评估重度肾功能损伤或未接受透析的肾衰竭患者(<30ml/min)的 PK、药效学、耐受性和安全性。建议设置足够长的随访期以评估药效学效应,该研究结果可指导后续试验的纳入/排除标准的制定。 如果药物靶向肝脏,建议评估肝功能损伤的影响。可在早期研究中序贯或适应性(sequential or adaptive)纳入不同程度肝功能损伤受试者,评估耐受性、安全性和药效学。上述信息有助于为后期试验中纳入不同肝损伤程度的受试者提供依据。 由于器官功能变化可能导致的药效学变化与 PK 变化不一致,因此在可行的情况下,应进行药效学评估。可基于群体 PK-药效学模型评估器官功能损伤与药效学、生物标志物、安全性或有效性之间的相关性。具体可参考 FDA Guidance《Exposure-Response Relationships-Study Design,Data Analysis,and Regulatory Applications(2003)》。除非有充分理由(如安全性考虑),整个开发计划应纳入足够数量的不同程度器官功能损伤受试者,以获得有意义的数据。 ◆ 案例分析: 在 Inclisiran 的临床开发过程中,通过 ORION-7(NCT03159416)试验考察了在肾功能损伤患者中 PK 和药效动力学。ORION-7 是 1 项在新西兰开展的单次给药、开放标签、平行研究,以评估肾功能损伤与肾功能正常受试者的 PK。该试验根据肾功能损伤的程度分为 4 组,分别为:肾功能正常组、轻度肾功能损伤、中度肾功能损伤和重度肾功能损伤。受试者在第 1 天接受 300mg Inclisiran SC 给药。主要终点为 PK 参数,包括 Cmax、Tmax、t1/2、AUC0-24h、AUC0-48h、AUC0-inf、SC 给药后的 CL/F 和 Vd/F、排泄分数、肾清除率(CLr)等;次要终点为 PCSK9、血脂、脂蛋白与基线相比的变化。 研究结果显示,在肾功能正常和损伤受试者中,Inclisiran 的药效学作用和安全性相似;肾损伤患者不需要调整 Inclisiran 剂量。虽然接受 Inclisiran 治疗的肾功能损伤患者的血浆暴露量更大,但对 LDL-C 水平、PCSK9 水平的降低程度和持续时间未见影响。该项研究提示 Inclisiran 可以安全地用于轻度、中度或重度肾功能损伤患者,且无需调整剂量或给药方案。 除此之外,还开展了 1 项 Ⅰ 期临床试验(ORION-6),这是 1 项单剂量、开放标签、平行研究,以评估肝功能损伤与肝功能正常受试者的 PK、药效学和安全性。结果显示,Inclisiran 可安全地用于轻度、中度肝功能损伤患者,无需调整剂量或给药方案。 详细内容可参考往期文章(点击链接:他山之石-4丨siRNA 药物 Inclisiran 上市历程及临床开发路径总结) 鼎泰集团内部总结 D. 药物相互作用的考虑 该指南讨论了评估 ONTs 与 CYP450 和转运蛋白之间潜在药物相互作用的考虑因素: 1. 与 CYP450 和转运体的 PK 相互作用 a. ONTs 作为 CYP450 和转运蛋白的底物 一般认为 ONTs 不经 CYP450 代谢,不太可能受到 CYP450 的影响。另外,对 P-gp 和 BCRP 等外排转运体,OATP1B1 和 OATP1B3 等肝脏摄取转运体,OAT1、OAT3、OCT2、MATE1和MATE2/K 等肾脏摄取或外排转运体的调节通常不会对 ONTs 的 PK 产生显著影响。 b. ONTs 作为 CYP450 和转运蛋白的调节剂 目前的经验显示,ONTs 对主要 CYP450 和转运蛋白的调节作用很小或没有。但不同类型的 ONTs 可能存在差异,仍需要逐一进行评估。 通常先进行体外评估,判断 ONTs 是否会抑制或诱导 CYP450 或转运蛋白。在进行体外试验和数据解读时,建议参考 FDA Guidance《In Vitro Drug Interaction Studies-Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions(2020)》。由于不同的体外系统之间存在差异,申请人应谨慎选择适当的体外试验系统。如果不进行体外评估,应提供合理依据。还应考虑 ONTs 通过其他机制(如干扰血红素或细胞因子的合成降解)与 CYP450 或转运蛋白发生相互作用的可能性。 如果体外研究显示 ONTs 可能调节 CYP450 或转运蛋白,应考虑开展临床药物相互作用研究。 2. 药效学相互作用 如一种药物的药理作用受另一种药物的影响(如作用机制相同的药物),ONTs 可能会与合并用药发生药效学相互作用。由于这种相互作用可能因具体药物而异,建议申请人就评估药效学药物相互作用的问题与 FDA 相关部门进行交流。 总之,由于 ONTs 与传统药物存在差异,在开发过程中需要全面评估其与其他药物在 PK 和药效学层面的潜在相互作用,以指导临床用药。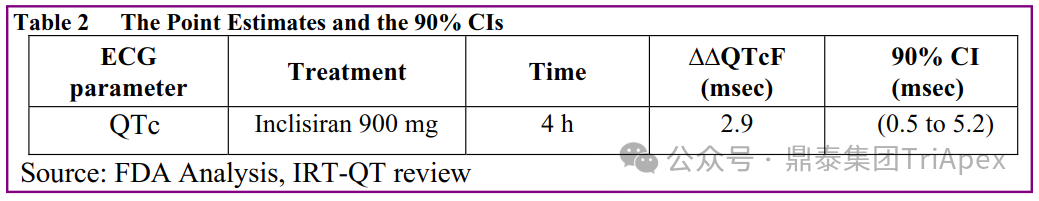


◆ 案例分析: 在 Inclisiran 的非临床研究中,开展了体外转运体试验,并根据研究结果及早期临床试验结果评估是否需要额外的 DDI 研究。根据试验结果, Inclisiran 不是 CYP 450 或转运体的底物、抑制剂或诱导剂,因此最终并未进行体内 DDI 研究。 典型的单次给药 DDI 研究可能无法为 Inclisiran 评估 PK 和 PD 相互作用提供足够的数据。在 EOP2 会议中,考虑到他汀类药物是拟开展的适应症中重要的联合用药对象,FDA 建议评估包括药效学相互作用在内的 DDI 的可能性。如在稳定状态下进行研究,如使用稀疏采样的方法;或者将 PCSK9 数据(含或不含他汀类药物)与他汀类药物 PK 一起收集,有助于和使用已有数据(例如 Ⅱ 期研究结果)来评估 PD 相互作用的潜力。 在 ALN-PCCS-001 研究中,比较了单独使用 Inclisiran(n=3-5)和 Inclisiran 联合他汀类药物(n=6)后 Inclisiran 的 PK 特征。结果显示,无论是在首次还是最后一次给药时,两组受试者的 Inclisiran 暴露水平相似。1 项 III 期临床试验(ORION-10)研究显示,Inclisiran 联合他汀类药物不会显著影响他汀类药物(阿托伐他汀和瑞舒伐他汀)的 PK 特征。上述研究显示,Inclisiran 与他汀类药物之间不存在显著的 PK 相互作用。 此外,考虑到临床上有联合用药计划,在毒理学研究中进行了联合用药的毒性评估,未发现对毒性或 TK 影响。 扫码下载 FDA guidance 原文
参考资料: [1] FDA Guidance:Clinical Pharmacology Considerations for the Development of Oligonucleotide Therapeutics(2024)https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-pharmacology-considerations-development-oligonucleotide-therapeutics [2] 鼎泰集团往期公众号文章







 法规政策
法规政策 项目管理
项目管理


 Global
Global



 苏公网安备32011202001081
苏公网安备32011202001081