


上一期,鼎泰集团转发了已上市的ADC药物的非临床研究的汇总整理(全球已上市ADC药物非临床研究一览-1)。为了更深入、系统地分析不同创新程度、不同适应症和作用机制的ADC药物的非临床开发路径,鼎泰团队系统整理并归纳总结了已上市ADC药物所开展的支持IND和BLA的非临床研究。 本系列分享按小分子毒素 (Payload) 将已上市ADC药物分为3类: 微管蛋白抑制剂,如MMAE、DM1、DM4 拓扑异构酶抑制剂,如DXd、SN-38 其他,如Calicheamicin 在研ADC产品payload的多样性及占比[1] 本期分享聚焦以微管抑制剂为Payload的已上市ADC药物(Kadcyla®、Elahere®、Adcetris®、Polivy®、Padcev®、Tivdak®等)的非临床研究历程(以MMAF为Payload 的 Blenrep®因确证性临床试验失败,未进行分析),同时结合CDE《抗体偶联药物非临床研究技术指导原则》(2023年)、ICH S9、ICH R6(R1) 和 ICH M3(R2) 对相关研究内容进行了分析。期望通过本次分享为业内同行ADC药物非临床研究提供精细参考,也期待与广大业内同行一起交流、探讨和提升。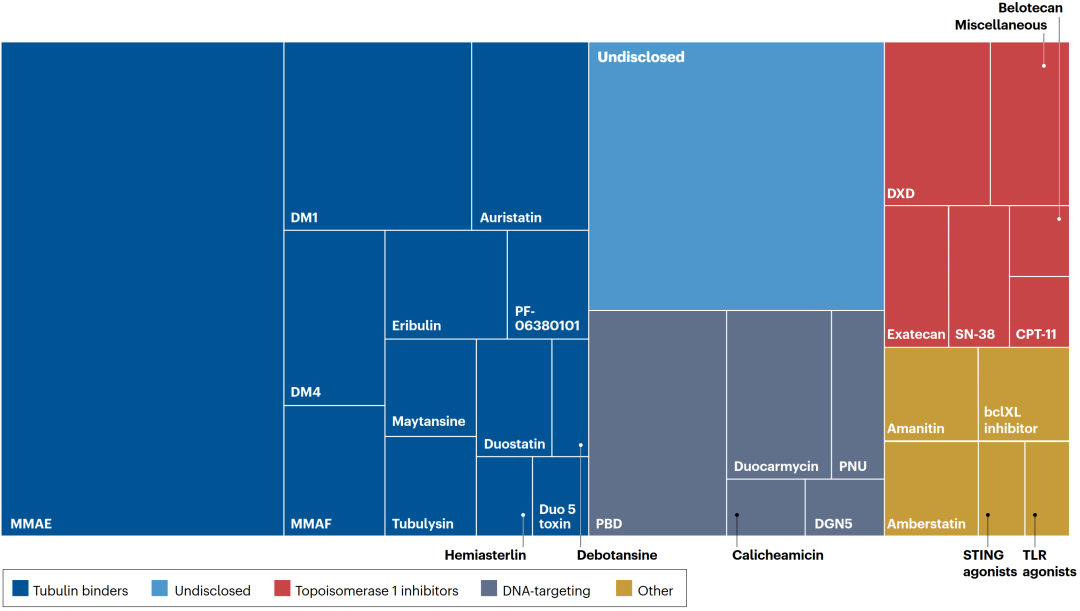
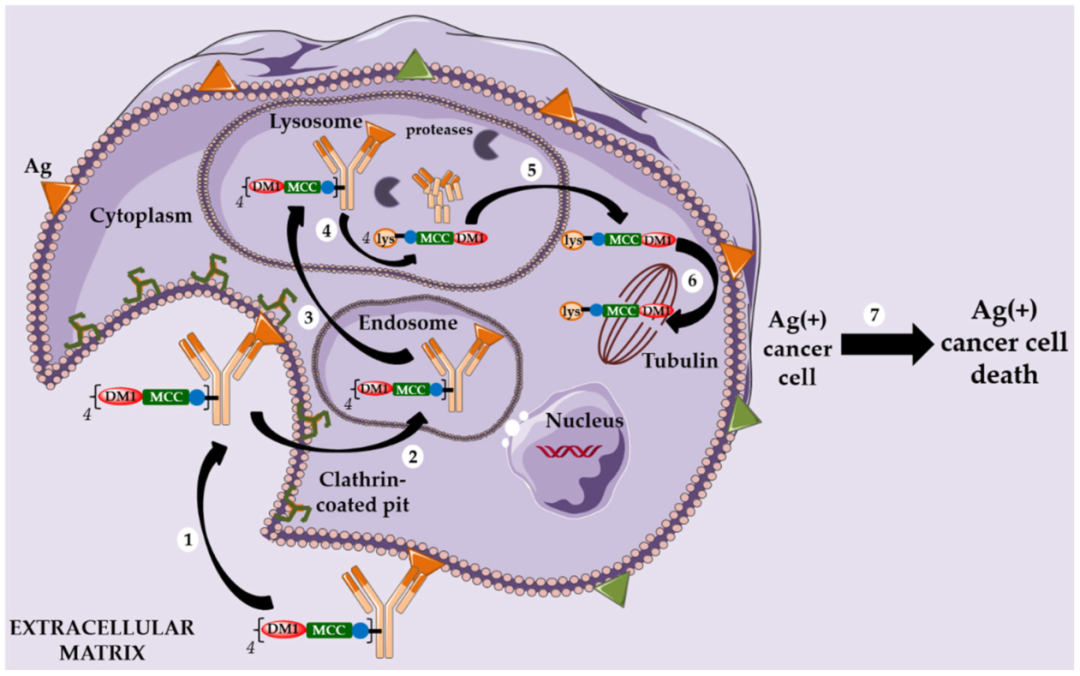
以微管蛋白抑制剂DM1为Payload的ADC药物的作用机制[2] 以微管蛋白抑制剂MMAE为Payload的ADC药物的作用机制[2]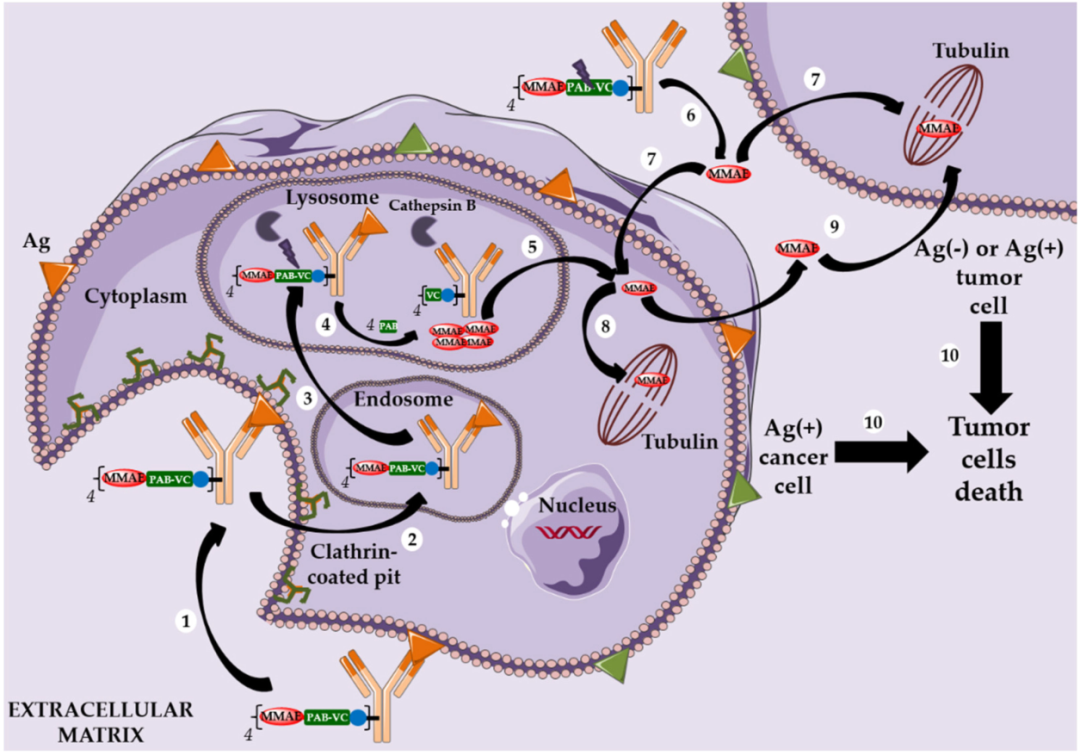
★ 目录 ★ 01 Kadcyla®(Trastuzumab emtansine)非临床研究内容 02 Elahere®(Mirvetuximab Soravtansine)非临床研究内容 03 Adcetris®(Brentuximab vedotin)非临床研究历程 04 Polivy®(Polatuzumab vedotin-piiq)非临床研究内容 05 Padcev®(Enfortumab vedotin-ejfv)非临床研究内容 06 Tivdak®(Tisotumab vedotin-tftv)非临床研究内容
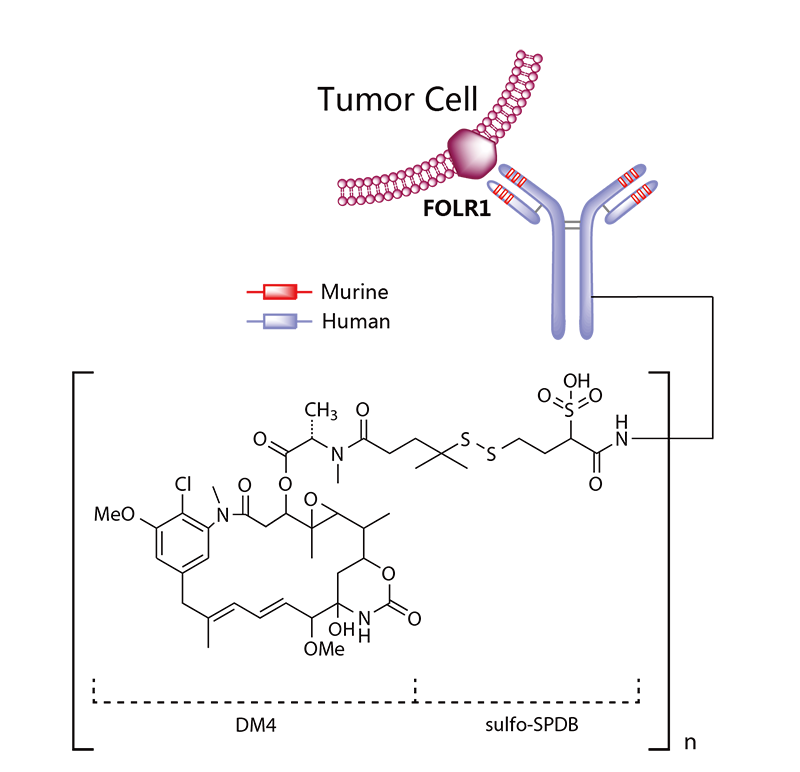
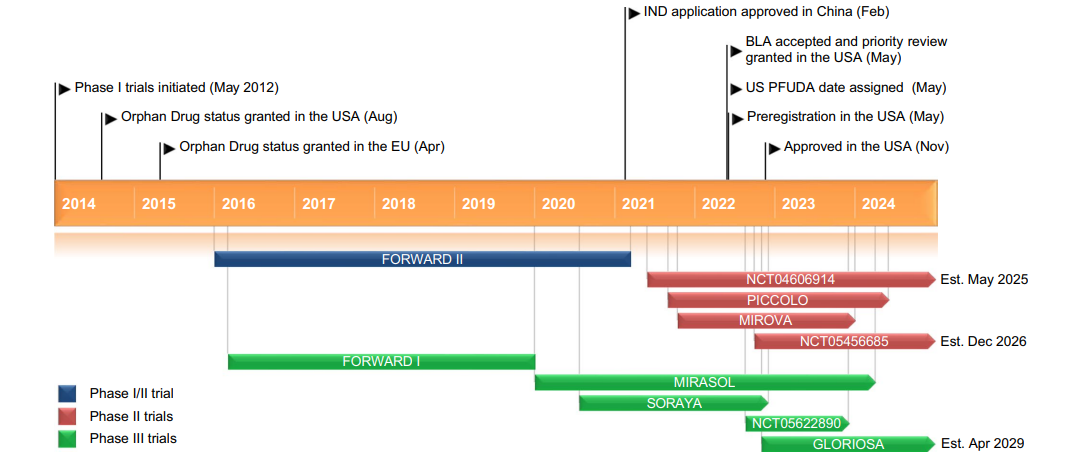
Kadcyla®(Trastuzumab emtansine) 非临床研究内容 通过不可裂解SMCC连接子将靶向HER2的抗体(IgG1)和微管抑制剂美登素衍生物DM1连接而成的ADC。 DAR约为 3.5,分子量约 148.781 kDa。 已获批适应症:用于治疗先前接受曲妥珠单抗和紫杉烷单独或联合治疗的HER2阳性转移性乳腺癌患者。 用法用量:临床推荐剂量为 3.6 mg/kg,静脉输注,每3周1次。 Kadcyla®(Trastuzumab emtansine)化学结构,图源药渡 Kadcyla®(Trastuzumab emtansine,T-DM1)开发的关键里程碑,图源药渡 2005年12月16日,向FDA提交IND申请。 2010年7月6日,提交BLA申请,被拒绝批准后于2012年再次提交BLA申请;2013年02月22日,由FDA首次批准上市。 | 支持首次人体试验(FIH)IND 申请的非临床研究 鼎泰团队自制图,点击查看更清晰 | 支持后续 IND 和 BLA 申请的非临床研究 鼎泰团队自制图,点击查看更清晰 由于DM1具有遗传毒性和对快速分裂细胞的细胞毒性,且Trastuzumab也存在生殖毒性,因此T-DM1的生殖毒性更大。回顾研究显示,6名接受Trastuzumab治疗的孕妇中有3名出现羊水缺乏症状[3]。结合 ICH S9 和 Trastuzumab 的生殖毒性试验结果,FDA认为上述研究足以支持T-DM1的上市批准。 Elahere®(Mirvetuximab Soravtansine)非临床研究内容 由抗FRα单克隆抗体(IgG1)通过可切割连接子与微管抑制剂美登素衍生物DM4连接组成。 DAR约为3.4,抗体分子量约150 kDa。 已获批的适应症:FRα阳性、铂耐药上皮性卵巢癌、输卵管癌或原发性腹膜癌。 用法用量:推荐剂量为 6mg /kg,根据调整后的理想体重(AIBW)计算,每3周静脉输注一次,直至疾病进展或出现不可接受的毒性。仅在5%葡萄糖注射液(USP)中稀释后静脉输注。 Elahere®(Mirvetuximab Soravtansine)化学结构,图源药渡 Elahere®(Mirvetuximab Soravtansine)开发的关键里程碑[4] 2012年5月启动1期临床试验。 2022年3月在美国提交BLA申请,2022年11月14日,获得加速批准上市。该上市申请基于如下关键性临床试验: 在一项单臂试验临床试验 0417(NCT04296890) 中,对106例先前接受过 ≤3 种治疗方案的FRα阳性、铂耐药复发上皮性卵巢癌、输卵管癌或原发性腹膜癌患者进行了疗效评估,所有患者均需接受过Bevacizumab治疗。主要终点为按照 RECIST v1.1 评价的ORR 和 DCR。结果显示,ORR为 31.7%(22.9,41.6),中位DCR为6.9个月。 | 支持首次人体试验(FIH)IND 申请的非临床研究 鼎泰团队自制图,点击查看更清晰 | 支持后续 IND 和 BLA 申请的非临床研究 鼎泰团队自制图,点击查看更清晰 根据ICH S9指南,晚期癌症适应症不需要开展致癌性研究。Elahere®的细胞毒性成分 DM4 破坏微管功能,具有遗传毒性,对活跃分裂的细胞具有毒性,这表明它有可能导致胚胎毒性和致畸性,未对Mirvetuximab soravtansine进行生殖和发育毒性试验。 Adcetris®(Brentuximab vedotin) 非临床研究历程 通过蛋白酶可切割连接子将靶向CD30的单克隆抗体(IgG1)和微管蛋白抑制剂单甲基澳瑞他汀E(MMAE)连接而成的ADC。 DAR约为 4,分子量约153 kDa。 已获批的适应症:自体干细胞移植后复发的霍奇金淋巴瘤(HL),以及复发或难治性全身间变性大细胞淋巴瘤(sALCL)。 用法用量:临床推荐剂量为1.8 mg/kg,静脉输注30分钟以上,每3周1次。 Adcetris®(Brentuximab vedotin)化学结构,图源药渡 Adcetris®(Brentuximab vedotin,SGN-35)开发的关键里程碑,图源药渡 2006年,启动首次人体试验(FIH),具体获得IND批准的时间未知。 2011年2月28日,向FDA提交BLA申请;2011年5月2日,获得优先审评;8月29日获批上市。该上市申请基于2项关键性临床试验: 在一项开放、单臂、多中心临床试验(NCT00848926)中,评价了Adcetris®对自身干细胞移植后复发HL患者的疗效。该研究共纳入102例患者,主要终点是客观缓解率(ORR)和缓解持续时间(DOR)。结果显示,接受治疗的患者ORR为 73%,中位DOR为 6.7 个月。 在另一项开放、单臂、多中心临床II期试验(NCT00866047)中,评价了Adcetris®对复发性sALCL患者的疗效。该研究共纳入58例患者,主要终点是ORR 和 DOR。研究显示,接受治疗的患者ORR为 86%,中位DOR为 12.6个月。 | 支持首次人体试验(FIH)IND 申请的非临床研究 鼎泰团队自制图,点击查看更清晰 | 支持后续 IND 和 BLA 申请的非临床研究 鼎泰团队自制图,点击查看更清晰 Polivy®(Polatuzumab vedotin-piiq) 非临床研究内容 通过蛋白可切割连接子(MC-vcPAB)将靶向CD79b的抗体(IgG1)和微管蛋白抑制剂MMAE连接而成的ADC。 DAR约为 3.5,分子量约150 kDa。 已获批的适应症:复发或难治性弥漫性大B细胞淋巴瘤(DLBCL)。 用法用量:临床推荐剂量为1.8 mg/kg,静脉输注90分钟以上,每21天一次,共6个疗程。 Polivy® (Polatuzumab vedotin-piiq)化学结构,图源药渡 Polivy®(Polatuzumab vedotin-piiq)开发的关键里程碑[5] 根据文献分析,推测在2010年前后提交IND申请。于2014年曾因严重的毒性反应而被叫停试验(partial clinical hold)。 2018年12月19日,向FDA提交BLA申请,并于2019年6月10日FDA加速批准上市,其基于1项关键的临床试验: 在一项开放标签、多中心临床试验 GO29365(NCT02257567),纳入了 80 名至少经过一线治疗的复发或难治性DLBCL患者,随机(1:1)接受Polatuzumab vedotin-piiq联合Bendamustine 和 Rituximab(P+BR) 或 BR,21天为一个给药周期,共给药6周期。Polatuzumab vedotin-piiq,在第1周期的第2天和随后周期的第1天静脉注射(1.8 mg/kg);Bendamustine在第1周期的第2天和第3天以及随后周期的第1天和第2天静脉注射(90mg/m2);Rituximab在每个周期的第1天静脉注射(375 mg/m2)。治疗结束时,P+BR和BR组的CR分别为 40% (95% CI: 25-57%) vs 18%(95% CI: 7-33%),ORR分别为 63% vs 25%。在25例对P+BR达到CR或PR的患者中,16例(64%)的DCR至少为6个月,12例(48%)的DCR至少为12个月。该适应症根据主要终点CR的结果获得加速批准。 | 支持首次人体试验(FIH)IND 申请的非临床研究 鼎泰团队自制图,点击查看更清晰 | 支持后续 IND 和 BLA 申请的非临床研究 鼎泰团队自制图,点击查看更清晰





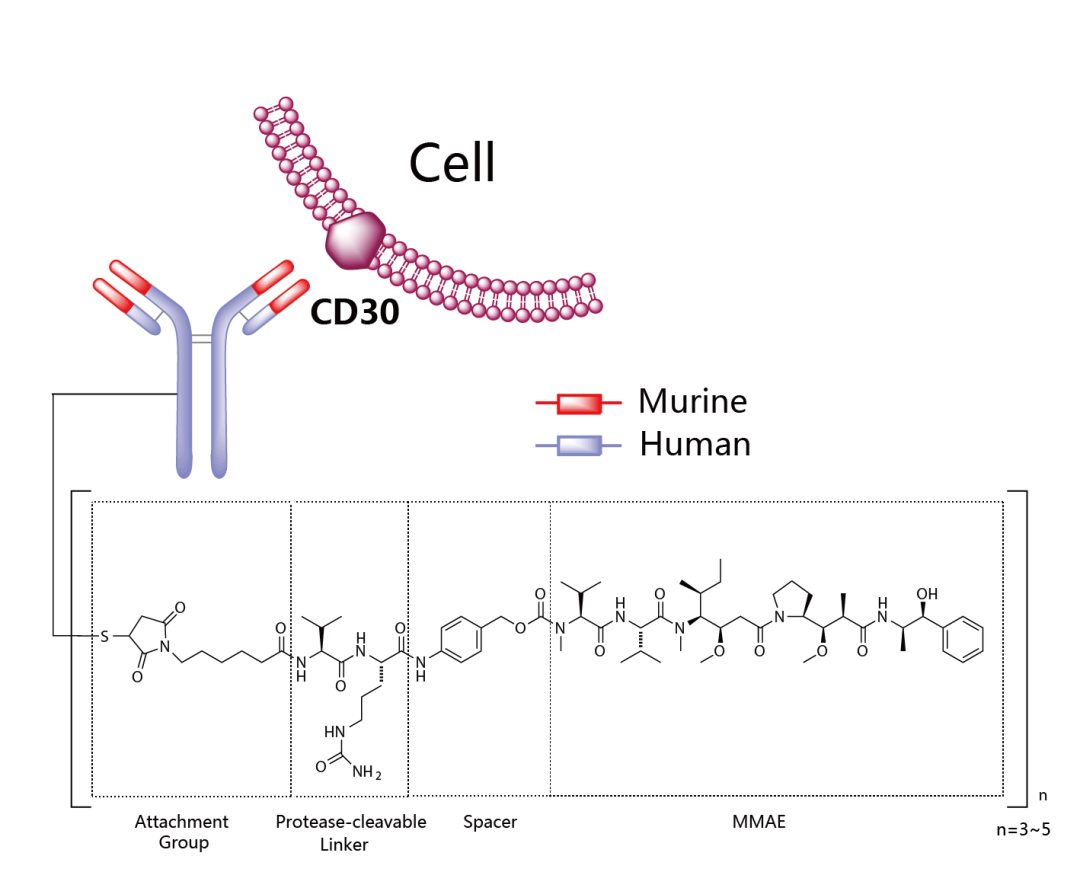





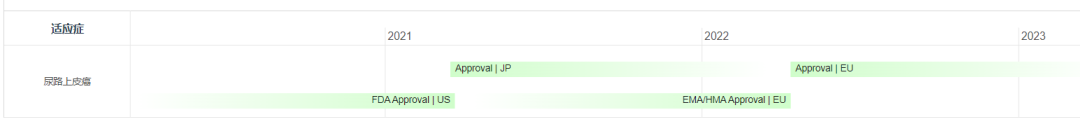
Padcev®(Enfortumab vedotin-ejfv) 非临床研究内容 通过马酰亚胺丙基缬氨酸-瓜氨酸(protease-cleavable maleimidocaproyl valine-citrulline, vc)连接子将靶向Nectin -4的抗体(Ig G1)和微管抑制剂MMAE连接的ADC。 DAR约为 3.8 ,分子量约152 kDa。 已获批的适应症:局部晚期或转移性尿路上皮癌,患者先前接受过程序性死亡受体-1(PD-1)或程序性死亡配体1(PD-L1)抑制剂,以及新辅助/辅助、局部晚期或转移性环境下的含铂化疗。 用法用量:1.25 mg/kg(最大剂量125mg),在28天周期的第1、8 和 15天静脉输注30分钟,直至疾病进展或不可接受的毒性。 Padcev®(Enfortumab vedotin-ejfv)结构,图源药渡 Padcev®(Enfortumab vedotin-ejfv)开发的关键里程碑,图源药渡 2012年12月28日,在FDA首次递交IND申请(IND116360)。在此之前,已启动代号为AGS-22M6E-11-1的首次人体试验(FIH)。 2019年7月15日提交BLA申请,2019年12月18日获得FDA加速批准上市。Padcev®(Enfortumab vedotin-ejfv)的获批上市基于如下关键性临床试验: 在一项单臂、多中心试验 EV-201(NCT03219333) 中,纳入 125 例既往接受过 PD-1 或 PD-L1 单抗和含铂化疗的局部晚期或转移性尿路上皮癌患者。在为期28天的治疗周期中的第1、8和15天给予 1.25 mg/kg Enfortumab vedotin-ejfv,直到疾病进展或不可耐受的毒性。主要终点为ORR和DCR。ORR为44% (95% CI: 35.1, 53.2),其中CR为12%,PR为32%;DCR为7.6个月(95% CI: 6.3)。该适应症的完全批准取决于确证性临床试验的结果。 | 支持首次人体试验(FIH)IND 申请的非临床研究 鼎泰团队自制图,点击查看更清晰 | 支持后续 IND 和 BLA 申请的非临床研究 鼎泰团队自制图,点击查看更清晰 小分子毒素MMAE的ADME部分试验参考 BLA 125388(Brentuximab vedotin),未单独开展,内容包括:MMAE的血浆蛋白结合试验,[3H]-MMAE的CYP酶代谢表型、[3H]- MMAE在大鼠、猴子和人肝细胞中的代谢产物鉴定和MMAE对人细胞色素P450诱导和抑制试验。MMAE的光安全性试验参考BLA 761121(Polatuzumab vedotin-piiq) 。 Tivdak®(Tisotumab vedotin-tftv) 非临床研究内容 由TF特异性人IgG1- κ单克隆抗体通过蛋白酶可切割缬氨酸-瓜氨酸(vc)连接子与微管抑制剂MMAE结合。 DAR约为 4,分子量约153 kDa。 已获批的适应症:复发性或转移性宫颈癌。 用法用量:TIVDAK的推荐剂量为2mg /kg(最多200mg),每3周静脉输注 30min,直至疾病进展或出现不可接受的毒性。 Tivdak® (Tisotumab vedotin-tftv)化学结构,图源药渡 Tivdak® (tisotumab vedotin-tftv)开发的关键里程碑[6] 2013年7月(GEN701)首次提交IND申请。 2021年03提交BLA,并于2021年09月20日 FDA加速批准上市。该上市申请基于如下关键性临床试验: 在一项开放标签、多中心、单臂临床试验(InnovaTV 204,NCT03438396)中,共纳入101例接受过不超过2种系统治疗的复发或转移性宫颈癌患者,且至少一种含铂化疗,69%的患者接受过Bevacizumab全身治疗。患者每3周接受2mg /kg的Tisotumab vedotin-tftv治疗,直至疾病进展或出现不可接受的毒性。ORR为24% (95% CI: 15.9%, 33.3%),中位DOR为8.3个月(95% CI: 4.2)。 | 支持首次人体试验(FIH)IND 申请的非临床研究 鼎泰团队自制图,点击查看更清晰 | 支持后续 IND 和 BLA 申请的非临床研究 鼎泰团队自制图,点击查看更清晰 小分子毒素MMAE的ADME部分试验参考BLA 125388(Brentuximab vedotin),未单独开展,内容包括:MMAE的血浆蛋白结合试验,[3H]-MMAE的CYP酶代谢表型、[3H]- MMAE在大鼠、猴子和人肝细胞中的代谢产物鉴定、 cAC10-vc-3H-MMAE或[3H]-MMAE在大鼠体内的排泄、物质平衡和药代动力学研究。 未针对 Tisotumab vedotin-tftv 开展胚胎-胎仔发育毒试验;MMAE(SGD-1010)胚胎-胎仔发育毒性试验参考 BLA 125388(Brentuximab vedotin)。根据ICH S9,拟用于晚期癌症患者的,可不开展生育能力和早期胚胎发育以及围产期发育毒性试验。 FDA根据申请人提交的 Brentuximab vedotin 申报资料(BLA 125388)对 Tisotumab vedotin-tftv 的遗传毒性进行了审评,包括:MMAE细菌回复突变试验(阴性)、MMAE小鼠淋巴瘤正向突变试验(阴性)、MMAE大鼠骨髓微核试验(阳性)。 结语 ADC药物的非临床研究需要考虑其复杂的组成结构和体内行为。抗体、连接子和小分子毒素三者之间的交互作用会影响药物的安全性和有效性。因此,非临床评价需要全面而审慎。 根据ICH M3(R2),开始支持FIH的IND申请前,进行ADC药物的血浆稳定性和毒素分子的血浆蛋白结合率试验有助于了解体内行为;针对性的体外和体内药效学试验有助于阐明ADC药物的初步作用机制、潜在的临床有效性,有时可为适应症的选择提供参考;对ADC药物进行短周期的重复给药毒性试验有助于获得支持临床试验设计的安全性数据。如果毒素为新分子,需要进行单独的毒性评价;如果该毒素分子有可参考的合法引用或公开的非临床数据,可借用文献资料(如MMAE)。在临床试验期间,可开展支持后续IND申请和BLA申请的更长周期的毒理试验,以及为解答前期非临床和临床试验中出现的毒性而开展的追加试验(如眼毒性);参考ICH S9,开展必要的生殖和发育毒性试验,对于拟用于晚期肿瘤患者的ADC药物,通常无需开展致癌性试验。在临床试验期间,可根据工艺变更的等级开展必要的非临床研究。 由于部分试验的起始时间在FDA审评报告中无法准确获知,鼎泰团队基于共识和经验进行了分析。如有不准确之处,欢迎同行提出宝贵意见,以提高该调研报告的严谨性。 鉴于篇幅所限,本文重点对多个已上市ADC药物的非临床研究路径进行了汇总和复盘,尚未深入分析每个产品的非临床策略和非临床研究的临床预测价值。后续鼎泰团队将选择代表性案例,从更为宏观的视角进行深度解读。这将有助于我们进一步理解和优化ADC药物的非临床研究策略,为后续同类药物的非临床研究提供借鉴,从而更好地支持临床试验设计和监管审评。 下一期,我们将分享以拓扑异构酶抑制剂为Payload的ADC药物的非临床研究内容,敬请关注。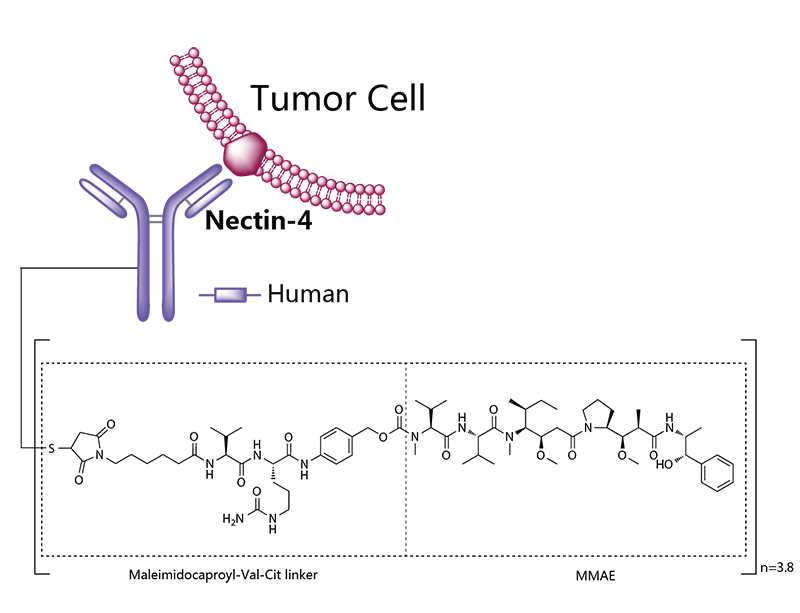






参考资料: 供稿:鼎泰集团转化科学和药政策略部







 法规政策
法规政策 项目管理
项目管理


 Global
Global



 苏公网安备32011202001081
苏公网安备32011202001081