


近年来,寡核苷酸药物新型递送系统的出现以及化学修饰技术的发展很大程度上改善了过去容易产生的脱靶效应和免疫反应,成为继重组蛋白药物、抗体药物之后的新一轮药物研发热点。寡核苷酸药物的分子大小介于传统的大分子和小分子之间,具有大分子和小分子的共同特征。这种模糊的身份引起了其免疫原性风险的争论。
目前已有14款寡核苷酸类药物上市,与其他生物制药类似,寡核苷酸类药物的免疫原性可能受到序列、给药途径、给药剂量、目标人群、共用药等因素的影响。目前已批准的不同寡核苷酸药物的抗药物抗体(ADA)数据表明,它们具有较低的免疫原性风险,且未发现对药代动力学(PK)、药效学(PD)和安全性产生重大影响。然而,在没有更多寡核苷酸类药物的数据之前,仍应谨慎评估其免疫原性。
本文简要综述影响寡核苷酸药物免疫原性的因素,以及评估时的考虑因素和分析方法检测策略。
深度学术文章
全文共7380字
预计阅读时间20分钟
寡核苷酸药物的分类和结构特点
寡核苷酸疗法(ONTs)是一种基于核酸的短合成分子,一般由12~30个核糖核苷酸或脱氧核糖核苷酸单链或双链组成[2][3],能够直接调控基因表达,被认为是继小分子药物和蛋白质类药物之后的新一轮药物开发方向。寡核苷酸(ON)药物的种类有很多,包括反义寡核苷酸(ASOs)、小干扰RNA (siRNA)、微小核糖核酸 (miRNA)、核酸适配体、免疫调节寡核苷酸等(表1)[3] [4]。ASOs、siRNA、 miRNA作用于细胞核或细胞质,而适配体与细胞外蛋白结合[5]。寡核苷酸药物的作用机制与传统的小分子和蛋白质疗法不同,其通过Watson-Crick杂交与细胞内同源RNA靶标结合,具有高选择性和高亲和力,从而形成异源的双链核酸分子。这些寡核苷酸利用内源性核酸酶降解靶RNA,或阻断宿主细胞内的核糖体机制调节RNA剪接和翻译[9]。
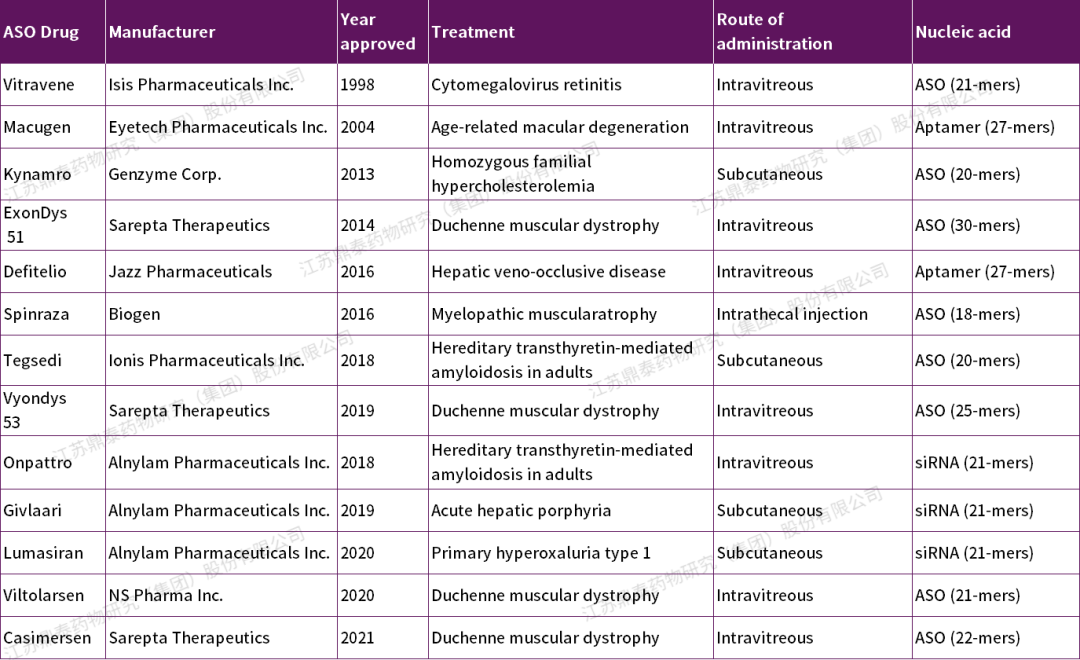
表1.FDA批准的寡核苷酸治疗药物[8]
寡核苷酸是一类独特的化合物,分子大小介于传统的大分子和小分子之间,具有大分子和小分子的共同特征。它们与大分子药物类似,具有相对较高的分子量,较长的半衰期和良好的靶点特异性。然而,它们也具有小分子的许多特征,包括同样是合成制备,化学结构相对简单,以及都会进行化学修饰以提高性能。这种模棱两可的身份问题引发了关于其相对免疫原性风险的争论。
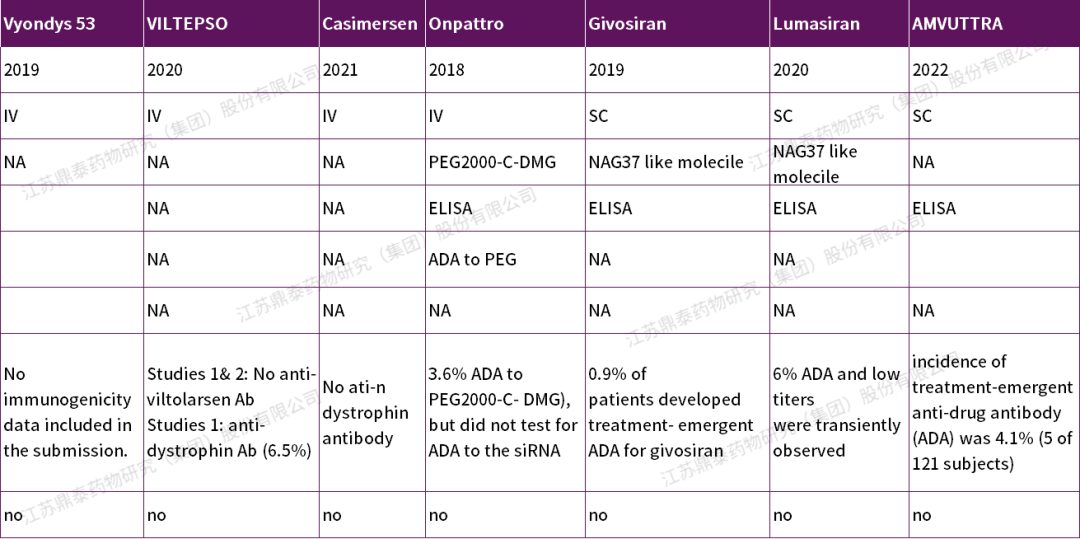
寡核苷酸药物通常会对杂环的碱基、核酸链,糖或二脂键进行修饰,这些修饰可能在外周引起免疫原性。但目前为止,当它们被运送到免疫限制的部位(如眼隙或中枢神经系统)时,对安全性的影响较小[1]。目前关于治疗性单克隆抗体和其他生物制剂(如生长因子)的免疫原性评估有相关的监管指南和行业白皮书已经发布[6][7],但缺少关于在寡核苷酸药物开发过程中寡核苷酸分子免疫原性的系统指南。(表2为已上市药物免疫原性情况汇总)
表2.左右滑动查看FDA批准的寡核苷酸药物的免疫原性情况汇总
蛋白质和多肽的免疫原性研究已较为深入和广泛,而对新型寡核苷酸类似药在动物或人体的免疫原性的研究报道还很有限。对寡核苷酸疗法的免疫原性的理解的局限,也限制了该类药物的研发,尤其是对免疫原性风险的评估。相信随着更多寡核苷酸产品进入临床和陆续上市而公开的免疫原性数据,这一情况会逐步得到改善。
影响寡核苷酸免疫原性的因素
01 产品因素
寡核苷酸是一种单链或双链DNA/RNA,分子量相对较小,与大分子治疗药物相比,不太可能产生抗药抗体(ADA)反应[9]。一般寡核苷酸与自然界中寡核苷酸具有某些相同的分子特征,可能使其免疫原性降低。然而研发过程中通常会对这些化合物进行化学修饰,以提高稳定性、安全性、细胞吸收及疗效。常见的修饰位点包括杂环的碱基,不同基团结合的核酸链,糖或二脂键进行修饰。这些修饰可能增加免疫反应[9]。免疫系统通常对内源性DNA/RNA具有耐受性,但会受到微生物病原体共有的一些碱基序列的刺激。因此某些新的寡核苷酸序列可能比其他序列的免疫原性更强。
寡核苷酸可以与其他类型的分子结合,如聚乙二醇、脂类、蛋白质或碳水化合物。对寡核苷酸治疗的非预期的免疫反应可能针对于载体、主干、寡核苷酸序列或任何由整个药物(载体加寡核苷酸)产生的新表位,因此评估其免疫原性应根据连接部分是否具有生物功能(例如:直接影响药效、PK、组织靶向的功能)、生物物理功能(例如:改变溶解度或稳定性的功能)或免疫修饰功能(例如:聚乙二醇或胶束形成的脂质本身可能具有免疫原性或阻止免疫系统与DNA/RNA类似物相互作用)进行具体分析。
02 给药途径
寡核苷酸可通过多种途径传递,例如皮下、静脉、脑脊髓膜内和眼内给药,其中皮下给药被认为具有较高的免疫原性风险。此外,迄今为止大多数已批准的寡核苷酸是递送至免疫限制或免疫赦免处,这些位点预期的免疫原性风险一般较低。
03 患者特异性
目前,大多数获批的寡核苷酸疗法主要用于受遗传疾病影响的受试者,而这些疾病与免疫反应强弱相关性较小。然而,人群的免疫激活(如自身免疫性疾病或炎症条件)、伴随治疗的药物 (可影响ADA的发生率或临床影响)等都会影响受试者。因此,未来的疾病类型可能与比目前发生的疾病具有更强的免疫原性相关性。除了这些因素外,目前ADAs是否对新的结构修饰或与原生寡核苷酸存在交叉反应风险,缺少临床经验和信息,但在未来预测寡核苷酸药物的免疫原性风险时,需要尽可能纳入考虑。
寡核苷酸类药物
免疫原性风险评估时的考虑因素
免疫原性的评价涵盖细胞免疫和体液免疫,本文主要是对于体液免疫相关免疫原性进行讨论。寡核苷酸疗法的免疫原性评估与常规抗体类药物类似,遵循FDA指导原则中的多层级免疫原性分析思路[7]。由于合成的寡核苷酸对人和试验动物的免疫系统可能同样是异源或同源的,因此,临床前的动物研究对预测人体中免疫原性具有一定参考价值。
寡核苷酸药物的免疫原性应基于风险进行评估。当存在针对单链或双链寡核苷酸药物(在某些自身免疫患者群体中尤其常见)或PEG(脂质纳米颗粒(LNPs)中的一种成分)或n-乙酰半乳糖胺(NAG37)(加入寡核苷酸以靶向肝细胞)的预存抗体情况下,测量细胞免疫原性和体液免疫原性,治疗前和治疗后,都能对免疫原性的评估提供参考[9]。
对于连接载体类药物,还应开发多种免疫原性测定方法来测量对寡核苷酸治疗不同成分的免疫反应 (例如,聚乙二醇化脂质纳米颗粒)或与蛋白质靶向配体偶联的寡核苷酸(例如,Fab片段) [9][10]。
一些寡核苷酸疗法的作用机制产生修饰的蛋白质(例如,剪接改变,外显子跳过寡核苷酸疗法)。在这种情况下,申办者也应考虑采用免疫原性检测方法检测修饰蛋白的抗体。此外,非预期的先天免疫激活也应在适当的时候进行检测。例如寡核苷酸治疗诱导的细胞因子释放,存在已知在人体中具有免疫原性的序列,如GU、CpG或5'-p,存在含有2'-deoxy、2'-OH或未甲基化胞嘧啶的天然核苷[10]。
01 关键试剂
在免疫原性分析方法中,阳性对照(PC)和药物偶联物是关键性试剂,试剂的亲和力、物种、亚型和特异性等特性均会影响分析方法的选择、开发过程中分析方法的优化以及验证过程中分析方法的性能。
由于寡核苷酸分子量较小,制备阳性对照和药物标记是免疫原性方法开发的最大挑战。通常将半抗原核酸药物通过固相合成的方法,在末端引入巯基,再加入马来酸酐类似物(马来酰亚胺)或NHS活化脂等,然后偶联至合适的蛋白(例如KLH或BSA等)得到全抗原;其次通过噬菌体展示或动物免疫制备阳性对照,调整免疫时间和次数以加强免疫;最终筛选获得高亲和性多抗或单抗用于ADA试验中的阳性对照。同样,由于寡核苷酸药物结构简单,无法通过后期偶联或者标记得到检测试剂,因此需要在合成过程引入活性基团或者常用的标记(Biotin,Digoxin等)。
02 样品采集
在整个临床项目中应采集ADA样本。如上所述,在没有预存抗体下,单次给药研究中可能没有必要检测抗寡核苷酸抗体。样品采集时间点必须与研究设计相适应。蛋白质和多肽类药物的样本采集时间点一般包括第一次给药前,给药后2周,4周,随后每3个月采集1次,最后时间点约为给药后的5个半衰期。这些时间可能适合首次人体寡核苷酸药物的研究。迄今获得的信息表明,给药后2周可能不是寡核苷酸药物ADA产生的有效时间点,部分寡核苷酸药物会发生延迟免疫反应。例如Tegsedi的临床研究(2018年批准)显示ADA的发生率很高(>30%),通常是发生在第一次给药后65周以上[12]。这表明长时间采样对寡核苷酸免疫原性的评估很重要。在临床Ⅰ期多次给药和剂量递增研究中不同时间点采集ADA样本,并用于临床Ⅱ期和Ⅲ期的采集时间点判断。同时建议早期开发中对样品进行储存(例如第Ⅰ期/首次在人体中的研究),如果有新的证据表明PK、PD或免疫介导的不良事件发生,再用于后续检测。
寡核苷酸免疫原性分析方法及检测策略
许多应用于蛋白质类大分子生物药物的生物分析方法同样可应用于寡核苷酸药物,但寡核苷酸药物独特的结构成分可能会给免疫反应和生物分析方法增加复杂性。
01 寡核苷酸药物ADA检测策略
一般来说,寡核苷酸药物遵循蛋白/抗体类产品的分层级免疫原性分析策略 (筛选、确证、滴度)。然而如果存在高发生率的预存抗体,可放弃单独的筛选试验,直接检测样品滴度,然后进行确证。
值得注意的是寡核苷酸药物一般与DNA具有类似结构。当药物进入血液循环系统,抗寡核苷酸抗体可能与内源性DNA发生交叉反应。有研究表明,狼疮患者的ADA可以与磷酸化寡核苷酸结合[11]。评估对寡核苷酸药物诱导的抗体对DNA的潜在交叉反应时,抗DNA测定方法的选择和优化至关重要。但目前为止,还没有建立合适的可用于筛选与DNA产生交叉反应的检测方法。
02 寡核苷酸药物ADA表征
给药途径和作用部位会影响产生的ADA免疫球蛋白(Ig)的种类,因此表征的检测需要针对性的考虑。血液样本的ADA检测应该能够检测到IgG和IgM亚型。如果作用部位在肠道或肺部,可以选择检测这些部位的IgA ADA。血清中IgA检测一般具有较优的方法一致性和灵敏度,也更容易被患者接受,通常作为分泌物IgA的代表检测物。IgE是另一种相关亚型。一些蛋白质生物制剂与严重的过敏反应有关。例如,ASO类似物ExonDys 51与患者的过敏反应相关,因此FDA要求对IgE ADA进行评估。
03 寡核苷酸药物中和抗体(Nab)检测
因为中和抗体会直接干扰药物靶向结合,因此,不希望有针对寡核苷酸药物的中和抗体产生。通常寡核苷酸药物靶点是在细胞内部,而ADA存在于细胞外循环系统中。阻止细胞吸收寡核苷酸药物的抗体会间接中和药物靶点结合,对药物靶点结合具有类似Nab的作用。考虑到细胞内作用机制,开发配体结合试验来确定ADA是否具有中和活性可能并不合适。
没有预存抗体的情况下,细胞内快速摄取寡核苷酸意味着ADA和Nab不太可能对单次给药或多次给药治疗的初始剂量产生任何影响。为了将寡核苷酸药物输送到特定的器官,一些寡核苷酸药物与细胞传递分子共价结合或与细胞传递分子合成复合物,因此对于寡核苷酸药物,Nab更多代表与细胞传递部分结合,以阻止药物传递到作用位点的抗体。对抗寡核苷酸药物的中和抗体需要一些特殊的考虑,这些考虑不同于蛋白质疗法,不同寡核苷酸药物也不同,需要更多考虑其结合和修饰方式。
目前尚未有针对上市寡核苷酸药物的中和抗体报导,但某些情况下监管机构要求上市后检测中和抗体(例如ExonDys 51和Defitelio)。我们建议将中和抗体检测纳入风险评估中,可以结合临床适应症在临床Ⅱ期或之后的试验中开展检测。
04 预存抗体检测
根据药物和患者人群的不同,一些受试者(例如RA,SLE或其他自免疾病受试者)在接受药物治疗之前可能会产生预存抗体。在某些情况下,预存抗体可能没有明显的影响。然而如果预存抗体能够清除药物,阻止靶细胞的传递/摄取或导致不良事件或产生针对双链DNA的抗体,并与寡核苷酸药物发生交叉反应,影响药品的PK、有效性或其他体内特性。在这些情况下,可能需要检测寡核苷酸和寡核苷酸的结合物(PEG、NAG37或其他部分)所产生的 ADA,并使用这些信息来确定受试者是否有资格参加临床试验。建议在Ⅱ期及之后的试验中对大多数患者人群检测针对双链DNA的抗体。如在患者人群中普遍存在,在Ⅰ期及之后的研究中检测。
免疫原性案例研究
已公布的寡核苷酸的免疫原性信息显示ADA发生率从非常低到很高(4%~72%),但对临床疗效或安全性的影响有限。目前已获得寡核苷酸的ADA数据表明,寡核苷酸都具有较低的免疫原性风险,对PK、PD和安全性没有发现较大影响。下面将介绍5个已批准药物的案例研究:
Onpattro™(patisiran) 是一种脂质复合物注射液,将siRNA包裹在脂质纳米颗粒(LNP)中,分子量约为14kD,静脉输注后递送至肝脏细胞内,靶向TTR信使核糖核酸(mRNA)以抑制TTR的产生。2018年获得FDA批准,用于治疗成人遗传性甲状腺转体介导的淀粉样变(hATTR),是首个获批的siRNA治疗药物。针对脂质纳米颗粒(LNP)的PEG2000-C-DMG成分,开发了一种ELISA检测方法:临床前9/60和25/60动物检测到抗PEG IgM和IgG;在安慰剂对照研究中,约4.1%的patisiran治疗患者(6/148)显示ADA阳性,但ADA滴度较低,且持续时间短,因此未进行中和抗体检测。数据显示未见ADA对Patisiran的临床疗效、药代动力学或药效学特征产生影响,属于低风险产品[13]。(方法学参数如下)
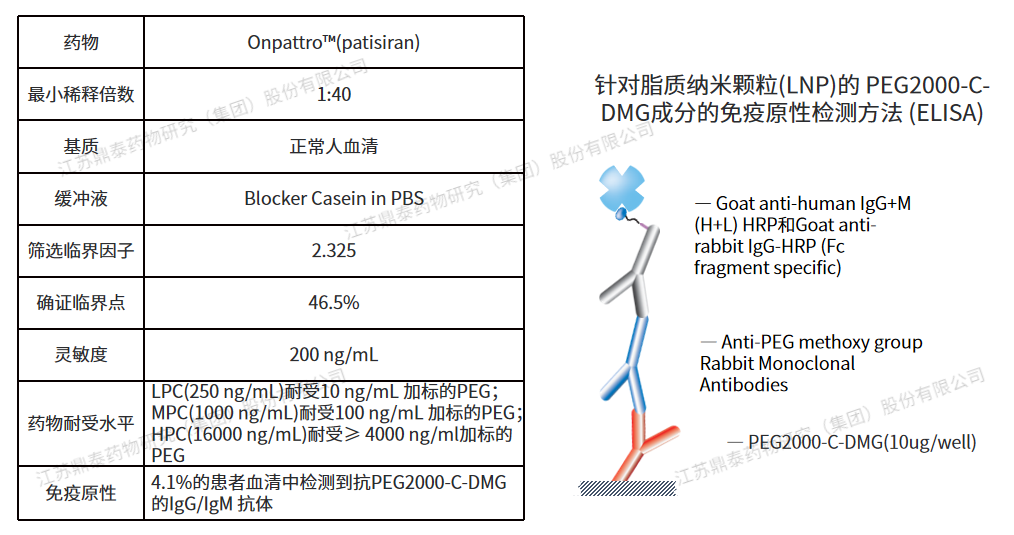
图1.Patisiran免疫原性分析方法及参数
Spinraza (nusinersen)是一种含有18个碱基残基的硫代反义寡核苷酸,可修饰SMN2基因的前信使RNA剪接,从而促进全长SMN蛋白的产生。2016年获得FDA批准,腰椎穿刺鞘内给药,用于治疗儿童或成年患者脊髓性肌萎缩症(SMA)。临床研究中共5例(4%)患者检测出ADA阳性,其中4例是短暂和1例是持久性的(滴度较低且呈现滴度下降),所有阳性样本的滴度范围为1至16。由于中和活性的主要风险来自高滴度持续的ADA,考虑到ADA的发生率、滴度和ADA的整体短暂性,因此未进行中和抗体检测。Spinraza寡核苷酸的独特结构具有诱导ADA与dsDNA交叉反应的可能,临床研究中未进行Spinraza对dsDNA的交叉反应性的评估。基于上述,FDA要求在上市后评估ADA阳性样品中dsDNA的交叉反应性。免疫原性检测方法采用过氧化物酶偶联的重组蛋白A/G为检测抗体, 蛋白质A/G主要结合人类IgG,因此实际的ADA发生率可能比报道的要高或发生得更早[14]。
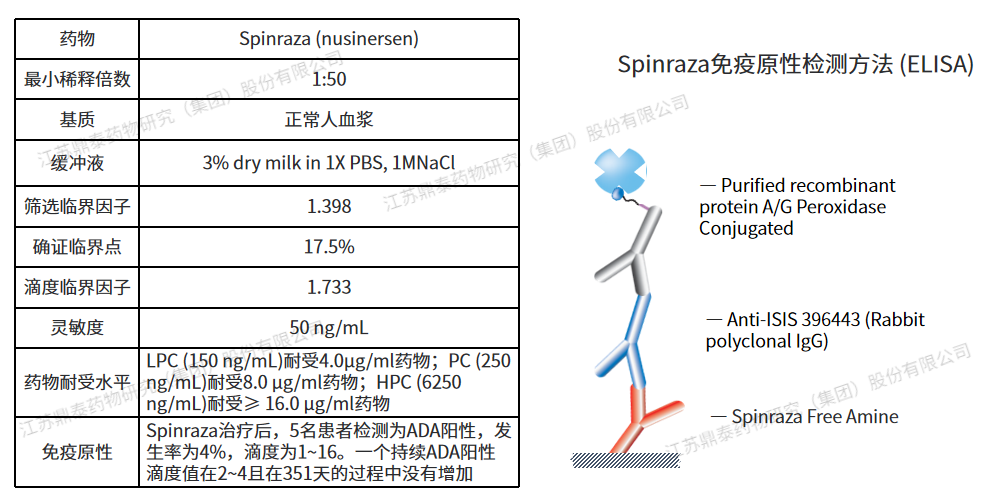
图2.Nusinersen免疫原性分析方法及参数
Givosiran是一种siRNA药物,通过正义链的3´端偶联到三价n-乙酰半乳糖胺(GalNAc),皮下注射递送至肝脏。2019年获得FDA批准,用于治疗成人急性肝卟啉症(AHP)。验证的ELISA法灵敏度为37.4 ng/mL,最小稀释倍数为50倍,在临床试验阶段检测抗Givosiran IgG和IgM滴度,未进行中和抗体检测。结果显示:约1%的临床研究患者发生了Givosiran治疗诱发的ADA,但未见ADA的产生对药代动力学产生影响,属于免疫原性低风险产品[15]。
ExonDys 51 (eteplirsen)是磷酰二胺吗啉代寡聚体(PMO)亚类的反义寡核苷酸,结构中包含30个碱基序列、39个吗啉环、1个哌嗪环。2016年获得FDA批准的用于治疗DMD基因突变导致外显子51跳跃的杜氏肌营养不良患者。在加速批准时,没有检测免疫原性,FDA要求上市后评估ExonDys (ASO) IgG和IgM,影响产品吸收中和抗体活性分析,过敏反应患者中IgE。目前这些免疫原性评估的结果还无法获得[16]。
AMVUTTRA™(vutrisiran)是一种化学修饰的双链小干扰核糖核酸(siRNA),靶向突变型和野生型转甲状腺素(TTR)信使RNA(mRNA),并与含有三个N-乙酰半乳糖胺(GalNAc)残基的配体共价连接,使siRNA能够传递到肝细胞。2022年6月获得FDA批准,每三个月皮下注射一次,用于治疗成人遗传性转甲状腺素蛋白淀粉样变性伴多发性神经病(hATTR-PN)。这是全球第5款获批上市的 siRNA 疗法。临床前研究中未见抗药抗体的对血浆中药物暴露产生影响。临床研究者中共有4例(2.2%)经vutrisiran治疗的受试者出现抗药抗体(ADA)。ADA检测阳性受试者中未观察到安全性、有效性、PK或药效学(PD)方面的显著临床差异[17] [18]。
结语
总的来说,考虑到目前常见的给药途径、药物的作用机制和目前已批准的寡核苷酸疗法的ADA数据,寡核苷酸药物具有较低的免疫原性风险,尤其是对于局部递送到免疫受限部位(如中枢神经系统或眼睛),在安全性和有效性方面的风险被认为更低。然而低免疫原性和低影响的报道可能受限于使用的分析方法和免疫原性监测的周期,因为寡核苷酸疗法诱导的ADA反应有时发展缓慢,可能为几个月而不是蛋白质疗法常见的1-2周。
因此我们建议在药物开发的早期进行寡核苷酸药物的免疫原性风险评估。在风险较低的情况下,开发的早期阶段收集和保存样本,并根据新出现的临床和暴露数据调查免疫原性,为后期和关键研究提供信息。迄今为止,虽然未发现与抗治疗性寡核苷酸抗体相关的不良事件,FDA和其他监管部门仍然强调对基因和细胞治疗产品的长期上市后安全的追踪。随着更多的寡核苷酸疗法进入临床,免疫原性风险及其可能的变化趋势,有待进一步观察。
参考资料:
免责声明:本文来自鼎泰集团内容团队,欢迎个人转发至朋友圈,谢绝媒体或机构未经授权以任何形式转载至其他平台,如需转载请添加微信LXL--7。本文仅作信息交流而非商业盈利之目的,内容仅供分享学习。







 法规政策
法规政策 项目管理
项目管理


 Global
Global




 苏公网安备32011202001081
苏公网安备32011202001081