


反义寡核苷酸类(ASO)药物通过Watson-Crick碱基配对规则与靶向的mRNA结合,可以治疗传统小分子或基于蛋白质策略难以实现的疾病。自第一个ASO药物Fomivirsen于1998年获批以来,FDA已经批准了10款ASO药物用于多种罕见病的治疗,并且还有更多的药物处于各个研发阶段。但在ASO药物研发的早期阶段,临床前研究侧重于改善药物的递送效率并评估其在靶组织中的分布和疗效,很少涉及对药物安全性特征的综合评估。
本文汇总了已上市ASO药物的临床不良反应,并通过对上述药物非临床研究结果的回顾,分析了非临床研究在指导ASO药物临床开发中的价值和局限性,探索非临床安全性评估中主要的考量因素,以期为后续ASO新药的开发与评估提供一定的参考。
已上市ASO药物临床不良反应汇总
根据分子作用机制,已上市10种ASO药物可以分为5类:
① RNase H-介导的mRNA 降解, 包括靶向CMV IE2 mRNA的 Fomivirsen、靶向Apo B mRNA Mipomersen和靶向转甲状腺素 (TTR) mRNA 的Inotersen;
② 诱导外显子跳跃药物,适应症均为肌营养不良,包括针对外显子51 跳跃的Eteplirsen、针对外显子53跳跃的Golodirsen、Viltolarsen和针对外显子45 跳跃的Casimersen;
③ 靶向运动神经元2 (SMN2)的pre-mRNA外显子修复的Nusinersen;
④ 通过与血管内皮生长因子(VEGF)受体相互作用的适配体药物Pegaptanib;
⑤ 针对多靶点的Defibrotide,通过与腺苷受体A1和A2结合,降低了纤溶酶原激活物抑制剂1(PAI-1)水平,具有抗血栓及纤溶活性。
ASO药物的作用机制和适应症各不相同,在临床试验中的不良反应也多样。一些不良反应属于ASO药物常见反应,如注射部位反应,头痛和发热等,这可能是由ASO药物的免疫反应所介导。Golodirsen、Viltolarsen和Casimersen可导致呼吸道感染引发咳嗽;而Defibrotide、Eteplirsen和Golodirsen存在ASO药物常见的胃肠道不良反应,如恶心和呕吐。常见反应如头痛、发烧、流感样症状、腹痛、恶心和疲劳,可以通过非处方药治疗。
已批准的ASO药物中,由于每种药物的吸收、分布、代谢和排泄(ADEM) 特性不同,寡核苷酸序列和化学修饰的不同,给药途径以及靶器官的不同,因此某些ASO药物具有独特的不良反应,如Fomivirsen和Pegaptanib可导致眼部炎症。
此外,需要重点关注的是FDA列出的黑框警告,如Mipomersen和Inotersen存在肝损伤;Pegaptanib、Defibrotide和Golodirsen出现超敏反应;Inotersen和Nusinersen可导致血小板减少;Golodirsen、Viltolarsen 、Casimersen和Nusinersen等大多数药物则存在明显的肾毒性。

表1. 已上市ASO药物的临床不良反应汇总
已上市ASO药物非临床研究毒性汇总
已上市ASO药物均在非临床研究阶段使用啮齿类动物(大鼠或小鼠)和非啮齿类动物(犬或猴)进行了不同周期的重复给药试验,同时也有部分药物就行了单次给药毒性试验。在给药方式上,除了局部给药的Fomivirsen、Pegaptanib和Nusinersen还存在额外的静脉输注外,其余药物在非临床阶段的给药方式均同临床给药。在非临床研究中常见的毒性反应是给药局部毒性和高暴露器官毒性反应,如表2所示。

表2. 已上市ASO药物的非临床研究毒性汇总
给药局部毒性反应常表现为眼内炎症、注射部位肿胀等,如Fomivirsen、Defibrotide和Golodirsen;最常见的系统性毒性表现为肝脏损伤和肾小管上皮细胞嗜碱性颗粒聚集和空泡化,如Mipomersen、Inotersen和Eteplirsen。此外,Viltolarsen和Casimersen等肾小管的改变和临床FDA列出的黑框警告是是密切相关的。其他毒性试验中,除Fomivirsen可能导致染色体结构畸变外,其他药物均没有遗传毒性;在致癌性研究中,大部分药物并未开展,在已开展致癌性试验中,Mipomersen可导致肝细胞腺瘤和皮肤/皮下纤维肉瘤的发生率增加。
非临床研究中的毒性特征与
临床不良反应的相关性分析
药物的非临床研究对于临床不良反应的预测至关重要,相关文献表明:同时使用啮齿类和非啮齿类动物的非临床研究对预测人体不良反应的阳性一致性为71%,单独使用非啮齿类和啮齿类的预测率分别为63%和43%[1],表明动物试验可反应大部分临床不良反应。
在已上市ASO药物中,大多数非临床毒性和临床不良反应是一致的。
如玻璃体注射(IVT)的两款药物Fomivirsen和Pegaptanib可导致眼部炎症;Mipomersen和Inotersen因肝损伤被列入黑框警告;Inotersen存在明显的血小板减少;Defibrotide在非临床试验中出现全身炎症反应增加,而在临床阶段则因过敏反应被列入黑框警告;Golodirsen、Viltolarsen 和Casimersen存在明显的肾毒性;Viltolarsen出现注射部位反应。
但也存在部分非临床毒性并未在临床中观察到的,如Mipomersen并观察到血小板减少;Defibrotide和Casimersen没有注射部位反应;由于人不同于试验动物,无法确定Nusinersen用药后是否导致神经元和胶质细胞坏死。
此外,部分反应因人和动物的差异以及检测方法的局限,无法在非临床试验中获得,如Fomivirsen和Pegaptanib可导致视力下降;Inotersen、Eteplirsen和Golodirsen未在动物中发现过敏反应;药物导致的各类疼痛、疲劳和恶心也无法在动物试验中检测到。
通过分析非临床研究和临床试验的不良反应,ASO药物的非临床毒性主要表现为免疫刺激反应,血小板减少以及高暴露器官(肝肾)的毒性[2][3]。由于ASO药物的核酸特性,在设计过程中,需通过化学修饰改善其稳定性,但化学修饰一定程度上可引起机体的免疫刺激反应,并且修饰后的硫代磷酸酯(PS)主链上的高亲和力化学物质可能会导致肝毒性或肾毒性。ASO药物一般外周达峰时间短,主要还是分布于相关靶组织及给药局部,易造成部分组织的药物高暴露。
ASO药物非临床安全性评估主要考量因素
结合ASO药物特性和不良反应表现,ASO药物的安全性评估考量因素可分为3类:(1)杂交和序列相关性因素;(2)杂交无关序列相关性因素;(3)杂交与序列无关因素。
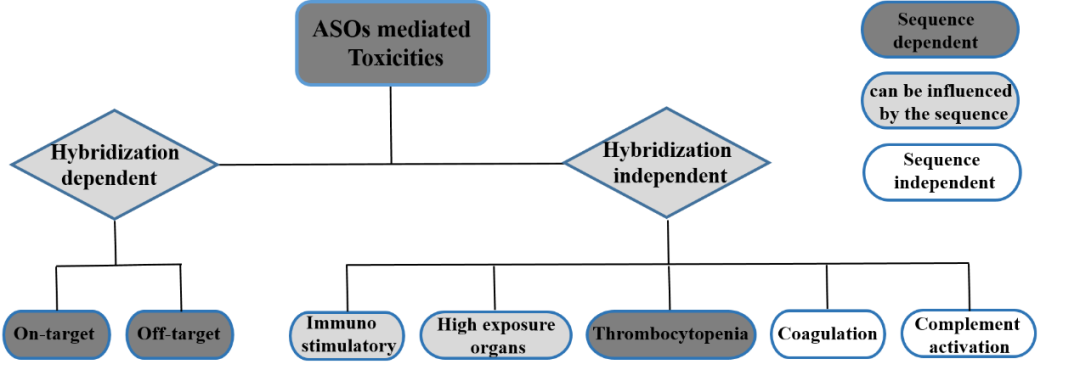
图1. ASO药物介导的毒性示意图
01 杂交和序列相关因素
杂交和序列均相关的毒性主要包括On-target靶向毒性和Off-target脱靶毒性。
靶向毒性是由ASO药物的药理作用放大而引起,可能导致过强的药理作用或在非预期组织中出现药物的不良反应。脱靶毒性则是ASO药物通过碱基配对原则作用于其他转录本而非预期转录本而引起的毒性[4][5]。
杂交和序列相关因素在药物设计时应被优先考虑,包括物种差异、化学修饰和生产性摄取等。脱靶毒性的评估可通过对靶向mRNA前体的整个转录组进行电子筛选以确定脱靶杂交的候选基因。靶向毒性的评估主要依赖于收集有关靶向的生物功能、组织表达模式的可用信息,以及来自同一靶向的其他药物的公开信息。
理想情况下,体外评估应在生物学相关的细胞系中进行,优先在人原代细胞或MPS中进行。体内评估需要在与候选药物具有物种交叉反应性的动物中进行。
02 杂交无关,序列相关因素
免疫刺激反应通常是ASO药物的主要反应之一,其毒性在很大程度上取决于核苷酸序列设计和化学修饰。ASO药物通过与TLR等模式识别受体(PRR)结合来激活先天免疫系统[6],在临床上导致注射部位反应、流感样症状和血小板减少[2],如图2所示。
免疫刺激效应的评估可在体外用分离的外周血单核细胞或全血测定预测细胞因子释放来评估。此外,有3款药物因临床出现瘙痒和荨麻疹等过敏反应而被FDA列入黑框警告,但在非临床研究中并未观察到过敏反应。对于非临床研究结果外推临床的提示性价值可能要考量种属免疫系统的差异性。基于核苷酸药物设计的不同,在ASO药物的非临床研究中,提前关注可能的过敏反应。

图2. 反义寡核苷酸介导的TLR途径
血小板减少在PS-ASO药物的非临床研究中较常见[7],如表3所示。
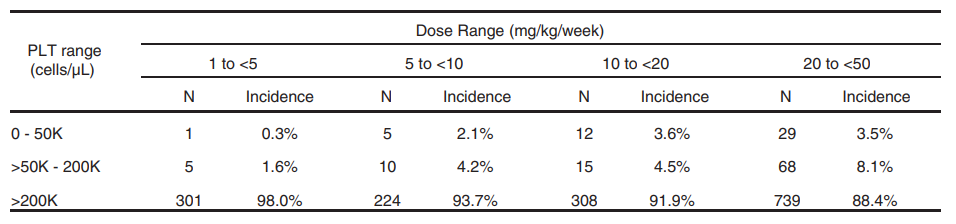
表3. NHPs在不同浓度ASO药物处理后
在指定范围内的PLT计数发生率
PS-ASO药物的NHPs的回顾性分析表明血小板减少症表现剂量依赖性,且一定程度上表现序列相关性。通过非临床与临床试验中血小板减少的转化研究表明,NHPs可以进行严重血小板减少症的风险预测,更多血小板功能的风险与控制及对血凝系统的影响仍需要基于临床研究数据判断。
血小板活化在体外可通过流式细胞术测定富含血小板的血浆中P-选择素(CD62P)和PAC-1(活化的GPIIb\/IIIa)的活化来确定。由于体外血小板活化仅代表潜在风险,临床上没有充分验证转化为血小板减少症,因此这些读数只能作为从具有不同效力的分子库中选择最佳小核酸药物的筛选标准。
基于ASO药物自身特性,在系统暴露后,药物通常在肝脏和肾脏出现蓄积。在非临床研究中,动物肝脏和肾脏出现细胞空泡化以及嗜碱性颗粒(被吞噬的寡核苷酸)属于ASO类药物常见改变,对非临床安全剂量的确定权重有限。但ASO药物导致的ALT和AST明显升高,则提示药物引起明显的肝损伤。
由于大多数ASO药物更倾向于通过肾脏清除,在超过一半以上的ASO上市药物中,肾小管出现了坏死,退化/再生等损伤,因此药物的肾毒性被FDA列为了黑框警告。究其原因,PS-ASO药物的肝肾毒性主要由其序列设计中更高亲和力的LNA和cEt等化学修饰核苷酸所引起[8],如表4所示。

表4. 不同序列和修饰的PS-ASO药物的肝脏毒性汇总
高暴露器官的毒性可通过体外预测模型以及检测早期毒性生物标志物来做临床前的安全性评估,此外,也可通过在不损失药物效力的情况下进行设计修改来降低ASO药物毒性。
03 杂交与序列无关因素
凝血功能的改变也是ASO药物不良反应之一。Nusinersen可见给药后的凝血异常。凝血抑制通常由达到阈值水平以上的血浆浓度驱动,其发生的主要原因是由于PS-ASO类药物的PS主链可通过抑制因子IXa磷脂复合物激活因子X或影响因子VIIIa的磷脂亲和力从而抑制凝血级联反应中的Tenase复合物(因子IXa和辅助因子VIIIa),进而选择性的抑制了部分凝血酶的作用时间,从而以一种与杂交和序列无关的方式抑制凝血反应[9][12]。
与凝血抑制类似,含有PS主链的ASO药物,如Mipomersen可与血浆补体因子H相互作用,降低了补体级联反应抑制剂的水平,导致级联反应不受控制的扩增。药物导致的补体过度激活可导致C3的耗竭,从而使补体功能改变,引发继发性炎症和血管炎[13][14],如图3所示。
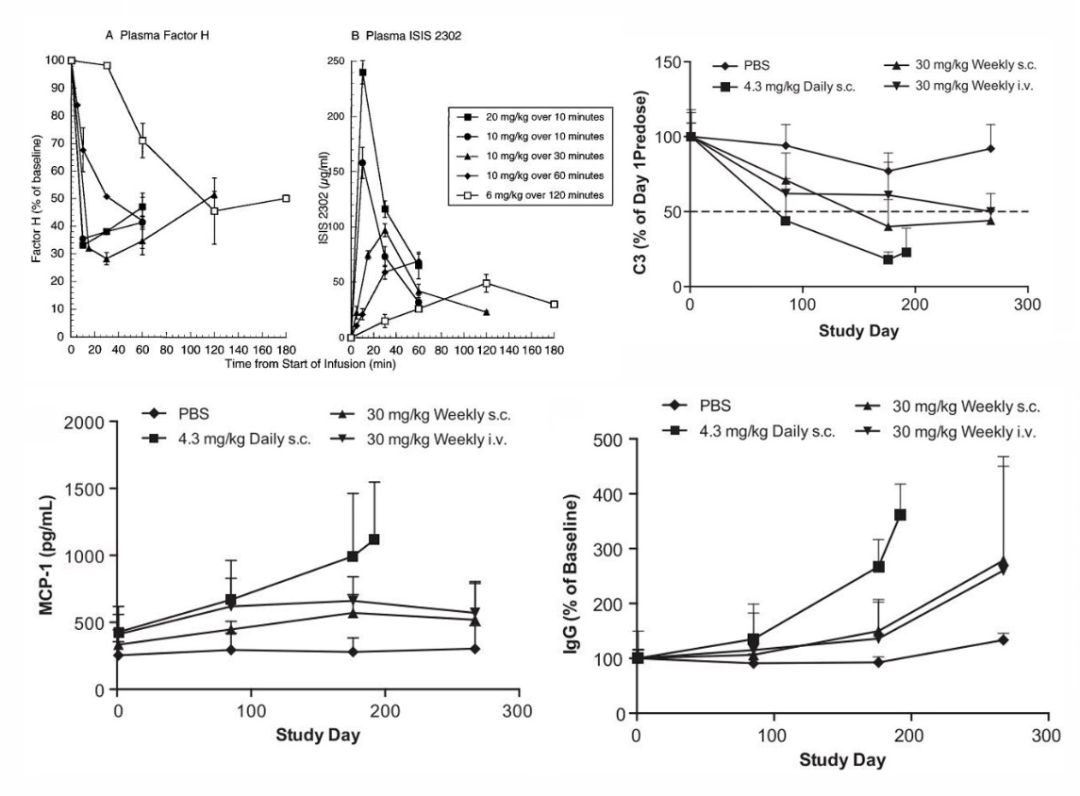
图3. ASO药物诱导的C3耗竭导致继发性炎症
凝血抑制可通过体外药物与柠檬酸血清孵育后测量凝血酶原时间(PT)和活化部分凝血活酶时间(APTT)来评估。而补体活化则可通过在小鼠、NHP或人的血清、血浆或全血中孵育候选药物后,用 ELISA测定补体途径(Bb、C3a和C5a)的不同分裂产物来评估。这类毒性在临床研究中并非常见,在大多数情况下,可以通过调整给药方案来减少对临床安全性的影响。
04 给药途径相关因素(中枢神经系统局部给药相关的毒性)
常用的中枢神经系统给药途径是鞘内给药(IT),在啮齿类动物模型中则是侧脑室内给药(ICV)。与全身给药相比,中枢神经系统给药总体给药剂量较低,因此导致的外周毒性风险也较低,但会导致脊髓液和大脑中药物浓度立即达到高浓度且持续较长时间,从而导致大脑诱导出现持续时间较长的免疫应答以及神经毒性。
这类毒性主要由ASO药物3’-鸟嘌呤核苷酸的数量和位置决定。现阶段神经毒性评估主要通过小鼠以及体外培养的原代神经元,但因为小鼠和人类神经元和非神经元细胞中表达的RNA存在显著差异,很难从动物模型中预测人体毒性,因此为了更好地预测ASO药物的神经毒性,还需要在动物和人类细胞模型中进行更多的研究。
05 其他因素
虽然当前对ASO药物引起肝毒性、肾毒性和超敏反应有了一些了解与推测,但在遗传水平上涉及毒性病变的精确分子和生化事件尚不清楚。如ASO药物引起毒性过程中发生的潜在分子事件是否影响其他小分子药物的ADME,改变其治疗效果,采用何种药理作用机制的药物可以降低该风险等。
结语
非临床安全性评估的目标是预测临床中潜在的安全风险。ASO药物的相关毒性主要取决于其序列设计、化学修饰、给药剂量和给药途径等。对于不同设计的药物,在临床前研究中需要从不同方面来考量和评估其安全性。
在已评估的3类因素中,杂交和序列相关因素在药物设计时应被优先考虑,杂交和序列无关因素则可在临床应用过程中通过调整给药方案来规避,而杂交无关但序列相关因素的担忧则需要在非临床及临床阶段受到重点关注。
到目前为止,大多数ASO药物都是用于治疗没有其他治疗手段的罕见性疾病,这使得药物的安全性风险考量在一定程度上让位于其治疗效益。但随着更多的用于常见疾病治疗的药物靶点的发现,更有效力和更敏感的体外模型的开发以及对不同毒性潜在机制的日益了解,对ASO药物的安全性评估将会更加严格,这也将进一步改进开发安全有效的ASO药物的过程,并促进ASO药物的更长远的发展。
参考资料:







 法规政策
法规政策 项目管理
项目管理


 Global
Global




 苏公网安备32011202001081
苏公网安备32011202001081