


17th生物分析领域最新问题研讨会 (WRIB) 于2023年6月19日至2023年6月23日在美国奥兰多举办,来自生物制药公司、生物科技公司,CRO 以及监管机构在内的1000 多名专业人员参与了此次会议。
本次会议的白皮书共分为3部分,涵盖约55个讨论主题,各部分内容将分别在《Bioanalysis》发表。

17th WRIB白皮书议题概览
鼎泰集团结合行业需求和团队既往经验,围绕最新发布的part 3内容,对相关议题进行了深入解读,汇总行业最新观点,以期指导药物非临床和临床阶段生物分析项的科学合理设计。
目前发布的17th WRIB白皮书 part 3 包括CGT、疫苗和生物制品的免疫原性两大部分,对包括dPCR、qPCR等技术方法,CGT与疫苗的免疫原性,CGT产品的PK和生物分布 (BD),生物制品ADA ISR、ADA方法开发等24个议题进行了分析讨论。本篇内容主要围绕这些议题对结论性观点进行汇总,以作参考。

17th WRIB白皮书 part 3议题概览
★ 文章导览 ★
01 | Section 1 Gene Therapy, Cell Therapy & Vaccines 关键词:dPCR;qPCR ;ELISpot;LNP;AAV;PK/BD;表达蛋白免疫原性; |
02 | Section 2 Immunogenicity of Biotherapeutics 关键词:ADA ISR;ADA 通用方法;复杂生物药免疫原性;临界值确定; |
03 | WRIB共识对当前生物分析的启示 |
04 | 鼎泰生物分析平台和技术能力 |
Section 1 Gene Therapy, Cell Therapy & Vaccines
基因治疗产品、细胞治疗产品与疫苗
| The Rise of dPCR
dPCR 的兴起
● 当缺少标准品无法进行绝对定量时,应考虑使用 dPCR 代替 qPCR,dPCR 在灵敏度、准确度和精密度上都优于qPCR;
● dPCR LLOQ 结合仪器 %CV 和 gDNA 含量确认,建议为 50 copies/μg DNA。
○ 建议将质粒片段加入全血或组织 (BD 试验) 中进行提取效率评估。
● 建议使用的定量单位:copies/ng DNA,copies/μL,copies/细胞:
○ 优先推荐使用 copies/ng DNA 和 copies/μL,如果使用对照基因进行了精密度、准确度和线性验证,也可以使用 copies/细胞。

dPCR平台的优势和注意事项[1]

dPCR平台验证建议[2]
| qPCR Assays Development/Validation
qPCR 试验的开发和验证
● 对 mRNA 进行 PK 分析时,选取的标准品应尽可能与药物原型接近 (药物本身或 LNP 包裹的 mRNA);
○ 需对标准品的稳定性进行评估。
● 目前核酸产品的 PK 分析平台有 bDNA,qPCR 和 dPCR;
○ 对于使用何种方法,监管机构并无要求,但不建议在临床前和临床研究之间更换检测平台,若更换需在 pre-IND 会议上与监管方沟通并获得书面的认可,并进行交叉验证。
● 临床研究中应注意检测方法特异性和灵敏度之间的平衡:
○ 对于药效研究,应保证特异性避免假阳性;
○ 对于临床诊断和流行病学检测,应保证灵敏度。
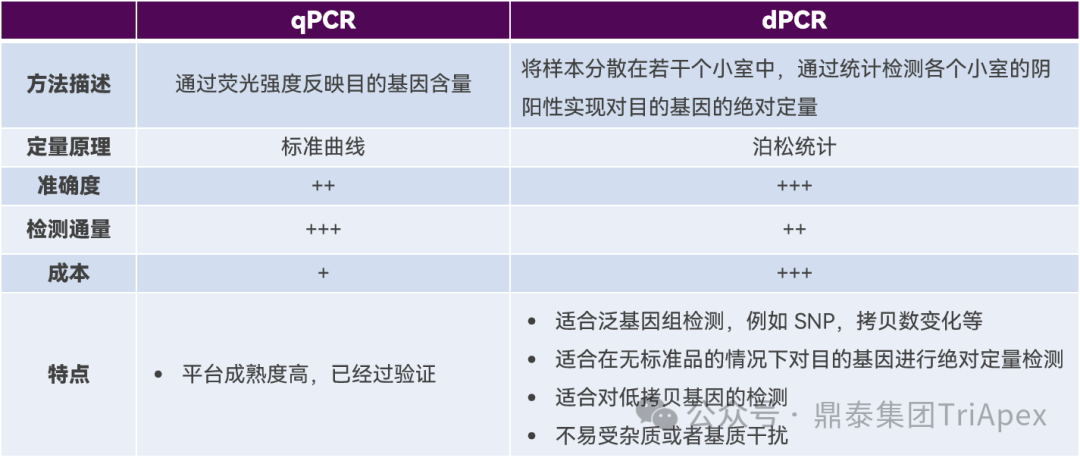
qPCR和dPCR平台选择考虑[3]
| Is ELISpot still the Gold Standard for Assessing Cellular Immunogenicity?
ELISpot 是否还是检测细胞免疫原性的金标准
● ELISpot 是目前广泛使用的细胞免疫原性检测方法,但有以下缺点:PBMC 的分离困难,样本需求量大,运输困难,细胞活力和功能易受影响,重复性差等。
○ 目前在样本制备和分析阶段有替代平台,如果 ELISpot 不适用,可以尝试开发验证替代平台。
● 建议在深入使用替代平台 (如 TruCulture) 之前,开展研究进行替代平台与 ELISpot 平台的对比;
● 关于细胞免疫原性检测的建议:
○ 对于自体来源的细胞治疗产品,可采集相关样本备测 (在出现安全性风险、PK 或其它细胞动力学参数改变时进行检测);
○ 对于异体来源的细胞治疗产品,应进行细胞免疫原性检测。
● 由于 ELISpot 的背景值变异度较高,不建议对免疫原性检测试验设定临界值。
| NanoString Assay Development/Validation
NanoString 试验的开发和验证
● 基于荧光条形码和单分子成像实现 RNA 绝对定量的高通量技术。自动化程度高,无需扩增也无需反转录,可对 >100 bp 或严重降解的样本进行分析;
● NanoString 验证试验的参数设置可以参考白皮书:
○ 应通过 RNA QC 样本和正式的稳定性试验评估 RNA 的降解情况进而评估 RNA 稳定性。
● 检测基因的数量和接受标准应根据试验开展阶段和 FFP (Fit for purpose) 准则设定,探索性试验可对部分基因设置接受标准,但进行伴随诊断 (CDx) 时需对所有基因建立标准。

NanoString平台验证建议
| NGS Assay Development/Validation
NGS 试验的开发和验证
● NGS 平台在检测 CGTs 的在靶效应和脱靶效应上具有重要的作用,但还需要大量的数据和经验支撑;
● 建议采用和 NanoString 相似的验证方法;
● 可以选择一些代表性的基因来确定检测灵敏度和测序深度;
● 如果检测广泛基因组改变 (例如基因组的插入和缺失突变),可以使用 wet lab 对照而非化学合成的对照作为阳性对照样本;
● 是否需要对具有技术挑战性的基因序列进行测序取决于药物本身的安全风险和计算机模拟或体外预测的基因突变频率:
○ 若具有较低的风险或较低的基因突变频率,综合评估后可以不进行深入分析;
○ 若具有较高的风险或较高的基因突变频率,应采用多种方法对相关区域进行测序。
| Issues with International Reference Standards for Vaccine Clinical Assay
疫苗免疫原性国际参考标准
● 疫苗免疫原性检测标准化是实现疫苗免疫原性评价一致性的关键;
● 疫苗免疫原性测定标准化是一项国际性工作,需要包括政府和非政府组织 (申办方)、标准实验室和工业界实验室的共同支持;
● 标准血清 (一级和二级) 是通过检测校准来标准化抗体浓度测定;
● 规范检测流程,使用合格的关键试剂对于标准化抗体浓度测定非常重要;
● 标准化策略的改变可能会对疫苗的研发造成影响,应在不同的检测实验室之间统一标准;
● 检测单位 (如国际单位 IU) 由所用的检测方法和所用的标准来决定。即使使用同一标准,也不能直接比较两种不同检测的结果。两个不同实验室之间的结果比较需要使用相同的检测标准和检测方法。
| Anti-AAV TAb Post-Dose Assessment: Development of an Efficient Analysis Strategy
给药后抗 AAV 总抗体 (TAb) 的检测:高效检测方法的开发
● 因为大多数给药受试者体内都含有大量的 AAV 抗体,因此 AAV TAb 检测结果对给药后 PK、PD 和安全性的影响评估具有局限性;
● 目前的研究数据显示 AAV 给药后的风险较低,但对给药后 AAV TAb 检测的必要性应基于不同项目的风险和现有数据进行综合评估;
● 对于系统给药例如静脉注射的 AAV 产品,为了提高检测效率,建议直接对部分样本进行滴度测定,剩余样本妥善保存;
○ 也可仅开展筛选试验,根据信号值区分阴性或阳性 (+,++,+++),不进行确证和滴度检测;
○ 也可只进行筛选和滴度试验但不进行确证试验,滴度试验可以进行 100 倍的预稀释,之后进行 5 倍或 10 倍的系列稀释至确定的稀释上限例如 100,000,最高的滴度值汇报为 >100,000,无需进一步稀释确认准确滴度;
○ 也可直接假定样品为阳性,不进行筛选和确证,直接按照第二种方法进行滴度检测。
| Further Considerations on LNP Immunogenicity
LNP 免疫原性的考虑
● 研究显示,PEG 抗体在人类中普遍存在 (高达76%),且预存 PEG 抗体的流行率正逐步增加,关于 LNP 免疫原性检测的方法开发和验证是否可以参照疫苗进行还需进一步讨论;
● 关于 PEG 抗体是否会对临床安全性和有效性造成影响还有待考究,可对 COVID-19 疫苗临床试验结果进行持续追踪。
● 可以与监管方沟通 PEG 免疫原性的检测方法,考虑使用其它检测模式:
○ 应根据风险评估结果确定检测模式和检测需求,无安全性和有效性风险可不进行 PEG 免疫原性检测;
○ 检测模式可以不遵循传统的分层检测方法,直接进行滴度测定;
○ 在开发新 LNP 疫苗平台时,应尽早进行风险评估以确定检测策略。
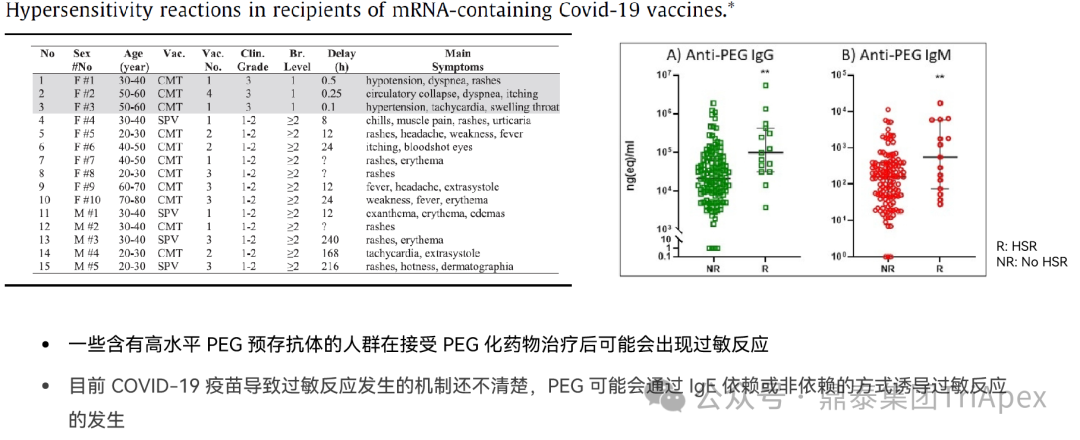
PEG免疫原性与COVID-19疫苗过敏反应的关系[4]
| Vaccine Clinical Assays Life Cycle Management
疫苗临床试验的全生命周期管理
● 疫苗的免疫原性检测应涵盖从临床检测到上市后研究,时间跨越较长,应有完善的全生命周期管理;
● 应根据试验目的 (如主要终点和次要终点) 确定方法转移过程中需要进行的桥接或交叉验证项;
● 如果方法改变程度小,可只进行桥接;如果方法已经搁置较长时间,应建立重启计划,必要时进行重验证;如果整个方法都被改变,则应进行全面验证:
○ 应使用可以覆盖整个试验范围的样本进行验证。
● 申办方应根据产品特性选择科学合理的检测方法,而不是一味遵从“金标准”。
| AAV TAb/NAb Assessment
AAV 总抗体/中和抗体 (TAb/NAb) 检测
● AAV NAb 方法开发应进行以下考虑:
○ 对 100+ 个体样本进行筛选是目前阴性混合血清制备的最可靠方法;
○ 对于 AAV NAb 的筛选和确证实验,建议使用统计学的方法确定临界值;
○ 为了减少 NAb 阴性血清的使用并提高滴度检测准确度,可先将样本进行中水平或高水平的预稀释,之后再按照 1:3 的稀释比进行梯度稀释;
○ 对于非临床研究 (例如 GLP 毒理实验,转基因表达蛋白的临床前 ADA 检测),NAbs 试验结果可提供的信息不多,TAb 检测结果足以支持对 PK 数据的解读,应基于风险评估决定是否开展。
● 若有需要可以在入组前进行伴随诊断 (CDx) 检测 AAV 抗体水平。
| Transgene Immunogenicity Assessment
转基因表达蛋白的免疫原性检测
● 建议在对转基因表达蛋白进行免疫原性检测之前进行免疫原性风险评估:
○ 检测前应考虑组织表达特征、剂量、给药方式、安全性和有效性风险:如果表达产物靶向特定组织,可能不需要进行系统的中和活性检测。如果是局部给药,或者目标蛋白为胞内表达,一般也无需考虑系统免疫原性。
○ 应妥善保存相关样本,基于风险评估和科学依据判断是否要对样本进行检测分析。
● 抗转基因表达蛋白抗体检测方法的开发需要综合考虑关键试剂 (替代蛋白、阳性对照) 、预存抗体或自身抗体,以免影响检测性能和临界值的确定;
● 关于免疫原性预测的计算机模型和体外工具的实际参考价值还有待探讨,使用目前通用的检测方法 (包括筛选、确证、NAb 滴度、5% 假阳性、1% 假阳性) 来检测转基因表达蛋白的免疫原性还有待进一步讨论,特别是低风险的产品。目前只有部分已上市的产品参照了蛋白药物的检测策略。
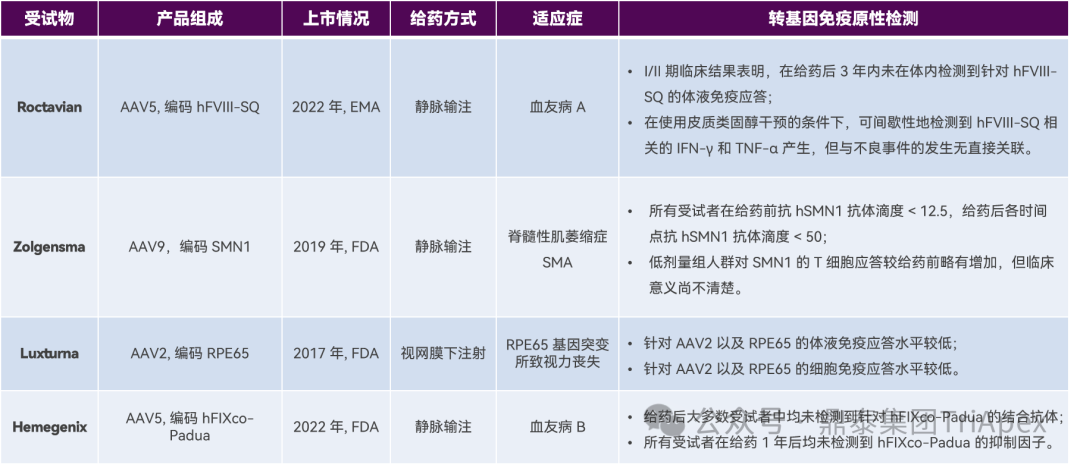
部分已上市药物转基因表达蛋白免疫原性检测结果[5]
| Vaccine Immunogenicity Assessment
疫苗免疫原性检测
● 疫苗检测试验的开发,确认和验证可参考 2020、2021、2022 年白皮书建议;
● 应基于科学评估确定检测类型和检测模式 (桥接还是 NAb) :
○ 对于 CDx 试验,总抗检测数据对临床更有用。
● 关于 singlicate 试验是否可以用于疫苗的免疫原性评估还未达成一致意见:
○ 建议获得更多的数据,特别对于滴度试验。
| PK & Biodistribution for Replication Competent Viral Vectors
复制型病毒的 PK 和 BD 检测
● 传统的分析方法例如 qPCR 不能对复制型药物和药物原型进行区分,可能会影响对 PK 数据的准确解读;
● 可以使用复制缺陷型工具病毒代替复制型病毒进行 PK 分析 ;
● 基于 ISH 的试验方法可以在细胞水平确定病毒载体药物的组织分布、定位和复制模式。
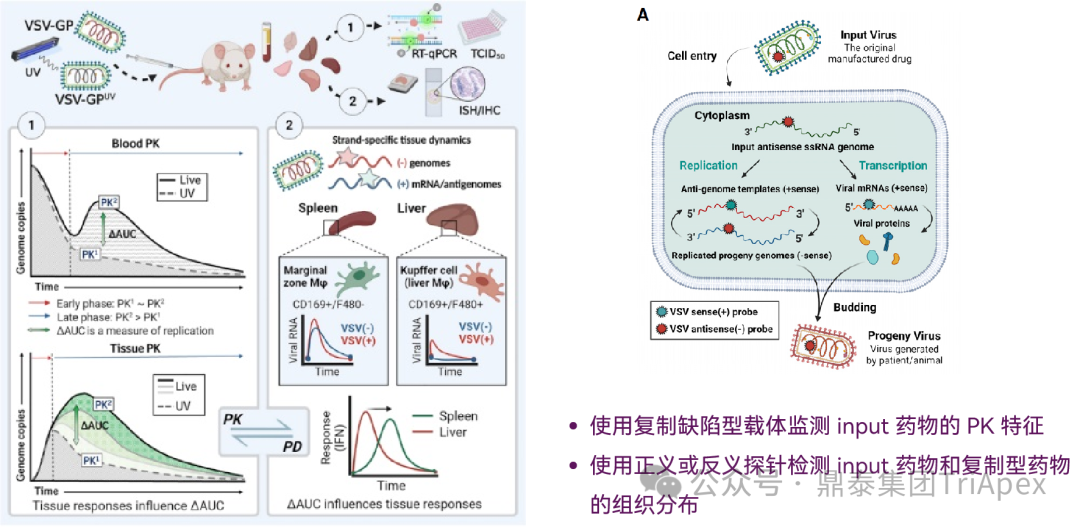
复制型药物与in put药物的区分[6]
| PK & Biodistribution for Virus-vector Vaccines
病毒载体疫苗的 PK 和 BD 检测
● 指导原则中提到需对复制型产品例如病毒载体疫苗开展感染试验,但也意识到目前感染试验面临如下挑战:变异度高、没有合适质控、组织匀浆的细胞毒性:
○ 综合评估使用最优方法。
● 需进行更多的讨论和数据支撑以进行进一步优化。
| PK & Biodistribution for CAR-T Cells
CAR-T 细胞产品的 PK 和 BD 检测
● 目前流式细胞术和 qPCR 是 CAR-T 细胞产品的主要检测平台,同时也有 dPCR 以及用于活组织检测的 IHC/ISH 的使用;
● 监管部门要求在出现不良反应时开展活组织检测 (继发癌变位点、有效性和安全性改变):
○ 此范围之外,是否进行活组织检查由申办方综合判断决定。
● 相比于 dPCR,FLOW 更适用于对细胞产品的监测。
Section 2 Immunogenicity of Biotherapeutics
生物治疗药物免疫原性
| ISR for ADA Assays: Reanalysis or Reproducibility?
ADA 试验的 ISR:数据再分析还是可重复性
● 在 ADA 的分层级检测中,一些样品可能至少经过两次检测,这些数据可以被用来评估已测样本的“可重复性”:
○ 提出了一种新型检测流程,即使用确认阳性的样本作为对照样本进行性能监测;
○ 这种新型检测流程可以被用来检测实验室内部数据的可重复性 (ISR),也可用于不同实验室之间的交叉验证。
● 一致认为筛选和确证实验的结果可以体现数据的可重复性,而且用可重复性”reproducibility”描述免疫原性检测更合适,不提倡对阳性样本进行再分析;
● 有建议提倡使用一系列临床样本监测检测性能的稳定性。
定量试验和免疫原性试验 ISR 的不同

| Strategies to Distinguish between Clinical PEA & TEA for Multidomain Drugs
区分多结构域药物预存抗体 (PEA) 和治疗相关抗体 (TEA) 的策略
● 对于多结构域治疗药物的免疫原性检测,可能需要开展除传统多层级检测之外的工作,例如结构域特异性检测和 PEA/TEA 的区分:
● 多层级筛选之外的检测工作需要开发特定的试剂和检测工具;
● 如果大量的 PEA 掩盖了临床疗效,应考虑使用生物工程替代药物将 TEA 从大量的 PEA 中区分开来。
| Universal & Generic Assays Formats for Immunogenicity Assessment
免疫原性检测的通用方法
● 非临床试验通常会涉及多种结构的药物,可以开发通用型方法来简化非临床免疫原性检测工作;
● 目前已经形成了多种可以使用通用试剂且具有较高药物耐受能力的通用检测方法;
● 非临床 ADA 方法验证和临床不同,可以简化非临床的 ADA 检测流程。
● 通用型的阳性对照可以用在非临床 ADA 检测试验中,是否可以应用到临床检测还需要根据相关数据进行进一步讨论。

| Immunogenicity Assay/Reporting Harmonization
免疫原性检测/汇报的统一
● 若使用 S/N 作为免疫原性结果呈现方式,应在首次临床试验期间验证 S/N 与滴度数据的相关性;
● S/N 的广泛使用需要得到大量数据的支持;
● S/N 相较于滴度值有较高的连续性,可作为新的免疫原性结果呈现形式;
● 当 ADA 数据被用来解释临床 PK 参数时,使用 S/N 比使用滴度更合适;
● 应提高 ADA 药物耐受和靶标干扰的浓度范围,以获得更多的试验信息,并满足监管要求;
● 尽可能增加验证试验的数据但也要控制试验开展时间;
● 目前免疫原性检测指南还远不能达到类似 ICH M10 的标准。

使用S/N汇报免疫原性检测结果案例[7]
| Immunogenicity Risk-based Approaches, Prediction & Mitigation: Focus on Novel Monitoring Strategies & in vitro Immunogenicity Assessment
免疫原性风险预测的方法:新型预测策略和体外评估
● 体外免疫原性风险评估方法是预测免疫原性的有利工具,有利于药物开发和生物候选药物的选择:
○ 在缺乏标准化方法和指导原则的情况下,需要对这些工具进行验证,以确保性能可靠;
○ 需要在公司内部以及行业内建立对照和基准。
● 可同时基于降解产物 (DP) 和副产物 (DS) 评估原料药 (API) 的免疫原性风险。API 变体的评估可以单独进行,然后比较相对风险;
● 当评估产品中的先天免疫反应调节杂质时,应该使用目标药物来进行评估,但同时要考虑不同制剂配方对检测的影响;
● 应尽早评估生物转化来为体外试验的优化提供更多时间;
● 当使用体外试验评估新分子的免疫原性风险时,建议首先筛查至少 10 名志愿者,以确定不同个体之间的差异,志愿者越多,后期筛选到合适候选者的概率就越大;
● 当在体外比较评估相关产品杂质的免疫原性风险时,所用细胞的 MHC 分布应能代表目标人群的 MHC 分布。一般而言需包括 30 ~ 40 名志愿者的数据。
| Lessons Learned from Immunogenicity Studies/Filings: Sharing Advanced Experience Related to Immunogenicity
Strategies/Approaches
免疫原性研究/申报的经验分享
● 不应比较不同分子间的免疫原性结果;
● 临床医师应认识到临床 ADA 的发生并不一定与临床反应相关;
● 若用 S/N 代替滴度结果,也应对临床医师进行相应指导;
● 使用单价重组 Fab 进行 ADA 检测的方法可以应用到其它治疗抗体的检测中,作为减少类风湿因子干扰的可行方法。
| Immunogenicity of Complex Biotherapeutics
复杂生物制品的免疫原性
● 申办方需要在早期临床实验中评估多结构域生物制品的免疫原性;
● 通常来讲,针对复杂生物制品 (例如双特异性抗体) 的免疫原性评估应检测不同结构域的结合特异性;
● 对于关键的临床研究,监管方期望可以对各结构域的免疫原性进行区分检测,但监管机构也可能会根据风险评估和安全指标按需调整指导原则的力度;
● 如果没有明显的临床反应,可以只汇报总抗水平,最好在前期与监管沟通,另外 NAbs 试验无需区分各结构域的免疫原性。
| Cut Point Assessment
临界值确定
● 目前关于样本假阳性率 (FPR) 的计算公式有多种,公认的方法是在整个公式中剔除确证阳性样本数 (分子和分母中同时剔除阳性样本),但如果阳性样本数较少,即便将其包括在总样本数中也不会对计算结果造成大幅改动,应在分析报告中说明使用的计算公式;
● 如果较难获得临床待测的基准样本进行临界值确定时,可以使用具有相似特征的受试者样本进行补充。
| Assay Comparability
试验可比性
● 关于 ADA 试验的可比性可参考 2021 和 2022 年白皮书,目前是使用已完成试验的 ADA 样品进行桥接来比较;
○ 需选择一系列样本来确定试验的可重复性,并应评估 ADA 的状态 (阳性/阴性) 以及应答程度 (滴度值或 S/N);
○ Cohen’s kappa 以及 MSR 对试验可比性评估具有重要作用;
●此外,临床和实验室标准 (Clinical & Laboratory Standards, CLSI) 指南侧重于诊断,可能在方法转移和验证的参数确定上具有参考价值。
WRIB共识对当前生物分析的启示
◆ dPCR, qPCR以及bDNA是核酸药物PK分析的常用方法,方法选择应Fit For Purpose。
◆ dPCR LLOQ建议为50 copies/μg;结果汇报单位建议为“copies/ng DNA,copies/μL”,特别情况下可使用“copies/细胞”。
◆ 疫苗免疫原性检测标准化是实现免疫原性评价一致性的关键,包括标准血清,检测流程,检测方法等的标准化。
◆ 对于基因治疗产品,免疫原性评估应包含细胞免疫和体液免疫。
◆ 应基于风险评估结果确定是否需要进行非临床AAV NAbs试验以及给药后AAV TAb检测。
◆ 对于复制型药物的PK和BD检测,应考虑在检测过程中区分In put药物和复制型药物。
◆ 目前流式细胞术和qPCR是CAR-T细胞产品的主要检测平台,特别是流式细胞术更适合对细胞产品的监测。
◆ 对于多层级的ADA检测流程,ISR更多体现的是检测的可重复性“Reproducibility”而非数据再分析。
◆ 对于多结构域治疗药物,应考虑对预存抗体、治疗抗体以及结构域特异性进行区分检测。
◆ 非临床ADA检测标准不如临床严格,可以考虑使用通用型阳性对照开发通用型检测方法,但其在临床检测中的应用还有待深入讨论。
◆ 使用S/N作为免疫原性汇报结果需有大量数据支持,需提高检测方法的药物耐受和靶标干扰耐受浓度范围。
◆ 当使用计算机模型或体外试验预估免疫原性时,需建立一致性标准对其进行验证,确保性能可靠。
◆ 应尽量使用可以模拟临床待测样本的基质进行方法开发,特殊情况下可以使用具有相似特征的受试者样本。
鼎泰生物分析平台和技术能力
鼎泰集团深耕生物分析领域10余年,构建了六大核心分析平台:免疫分析、细胞分析、qPCR/dPCR、LC-MS/MS及LC-HRM、超灵敏分析以及多因子检测等。我们的生物分析平台已成功助力700+项目的顺利交付,在常规项目中我们精益求精,追求卓越;在siRNA类、AAV类、疫苗类、ADC类以及其他创新型产品的生物分析项目实践中,我们也积累了丰富的经验,展现了专业实力。我们拥有1000+生物分析方法开发与验证经验、建立了200+关键试剂库、并开发了100+即用型分析方法、此外我们还拥有丰富的生物资源库(包括基质、组织等)。为各类型药物的临床前和临床试验提供全面的样品分析服务。
鼎泰生物分析始终从监管要求出发、秉持科学原则、紧密追踪客户需求。我们注重技术创新,积极储备资源,致力于提高分析效率和响应速度。经过本次对WRIB白皮书part 3的学习,我们对生物分析的现状和未来有了全新的认识和思考,也对当前我们自身的知识体系和技术能力提供了科学性指导。我们将持续关注行业动态,不断学习和提升,同时也热切期望能与业界同行进行更多的探讨、交流和学习。

往期文章精选
1、ICH S12《基因治疗产品非临床生物分布的考虑》要点汇总(上)
2、ICH S12《基因治疗产品非临床生物分布的考虑》要点汇总(下)
参考资料:







 法规政策
法规政策 项目管理
项目管理


 Global
Global



 苏公网安备32011202001081
苏公网安备32011202001081