


上期文章,鼎泰集团先后对临床试验失败的6款siRNA药物及失败原因(点击查看原文)、已上市的6款siRNA药物的非临床药效学内容(点击查看原文)进行了复盘分析,受到了同行的广泛关注。随着siRNA 药物的快速发展,质量控制和确保临床用药安全成为行业关注的焦点。
本期,我们将切换视角,对siRNA药物杂质水平控制策略进行调研和梳理,以期为siRNA药物的研发提供参考。
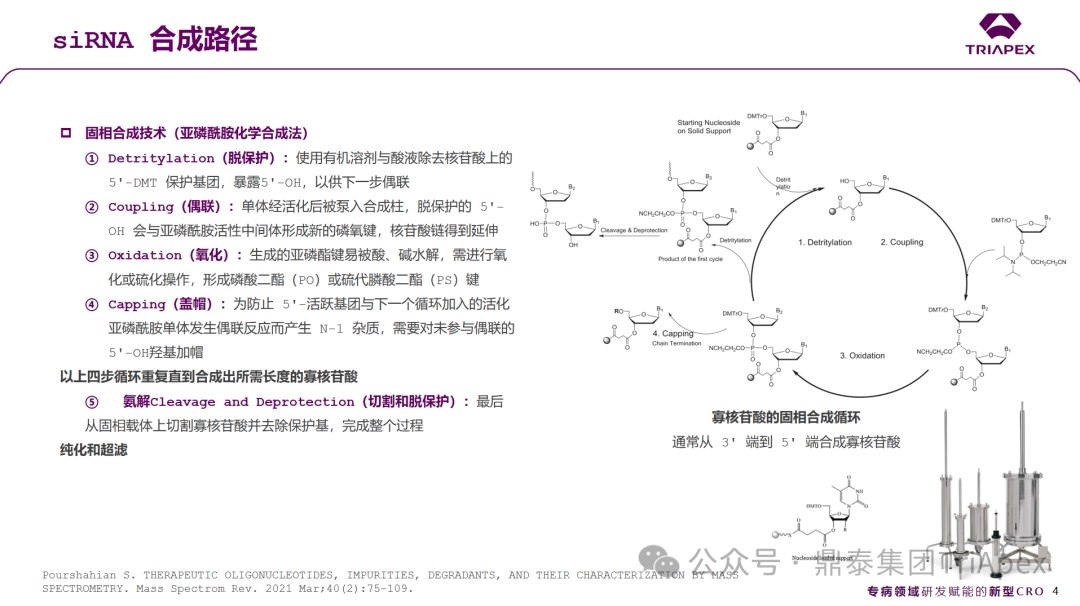
siRNA 药物合成路径和杂质来源
与小分子化学药物不同,siRNA 药物具有更大的分子量和更复杂的化学结构,其研发和生产过程中产生的杂质也异常复杂。如何科学评估siRNA药物杂质的影响和合理控制其限度,是业界亟待探索的重要课题。
众所周知,siRNA主要是通过固相合成(solid-phase synthesis,SPS),常伴随产生多种工艺相关(如起始原材料、中间体、配体和保护基团残留等)或产品相关杂质(与活性成分结构接近的副产物),这些杂质可能会对siRNA药物的质量和安全性造成潜在影响。
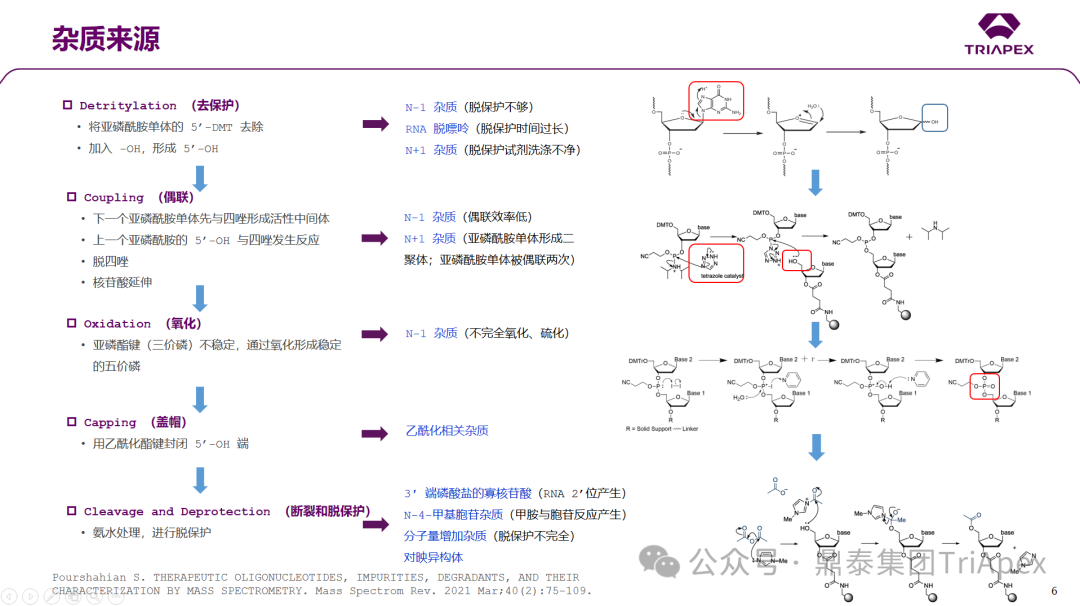
siRNA 药物杂质研究遵循的指导原则
siRNA药物相关的有机杂质、无机杂质、残留溶剂和元素杂质等可遵照ICH Q3系列指导原则;考虑到与常规化学药物之间的差异,siRNA药物产品相关的杂质的报告限度(Reporting threshold)、鉴定限度(Identification threshold)和界定限度(Qualification threshold)不能完全按照ICH Q3A确定。因此,对siRNA药物各类杂质水平的科学控制尤为关键。
siRNA 药物杂质研究的考量因素
1
对于杂质研究的化学考量
◾ 杂质表征
对产品相关杂质的表征是任何新化学实体开发过程中的一项重要工作,需以对合成工艺、起始物料和潜在降解途径的详尽评估为指导。由于siRNA 药物的平台特性,基于一个分子上获得的信息可用于指导其他分子的杂质研究,减少重复性工作。对于已有充分文献报道的杂质,对表征研究的要求较低;对于首次报道的杂质,则需要更全面的数据支持其表征研究。对于由多个成分构成的混合杂质,通常不需要逐一解析不同成分的结构。有一句老话 “A day in the library saves a month in the laboratory”用来形容siRNA 药物杂质研究特别贴切。
◾ 分析方法
应尽可能开发能有效分离杂质与siRNA药物序列以及不同杂质的分析方法,如毛细管凝胶电泳法(CGE)、阴离子交换色谱等。近年来,离子对高效液相色谱-电喷雾电离质谱联用(IP-HPLC-ESI-MS)技术的应用日益广泛。对双链siRNA药物,除了分析单个单链外,还需要在非变性条件下进行分析,以将残留单链和聚集体与siRNA药物双链进行分离和定量。
◾ 杂质报告
与小分子化药一样,siRNA 药物的质量标准中也需列明特定杂质和非特定杂质。前者有助于评价生产工艺中特定步骤或控制策略的成效,如单链寡核苷酸通常含有较低水平的 n - 1 杂质,其水平在很大程度上取决于固相合成工艺中脱苯甲基步骤是否成功。考虑到siRNA 药物杂质难以全部有效分离,特定杂质常需要以组的形式报告,如将n-1或n+1的所有组分归为一组。
对于大多数小分子药物,报告限度设定为0.05%或 0.03%。鉴于分离和分析方法的局限性,目前尚没有形成广泛接受的寡核苷酸药物的报告限度、鉴定限度和界定限度。在大多数情况下,设置0.2% 的报告限度、1.0% 的鉴别限度和1.5%的界定限度可能是适用的。
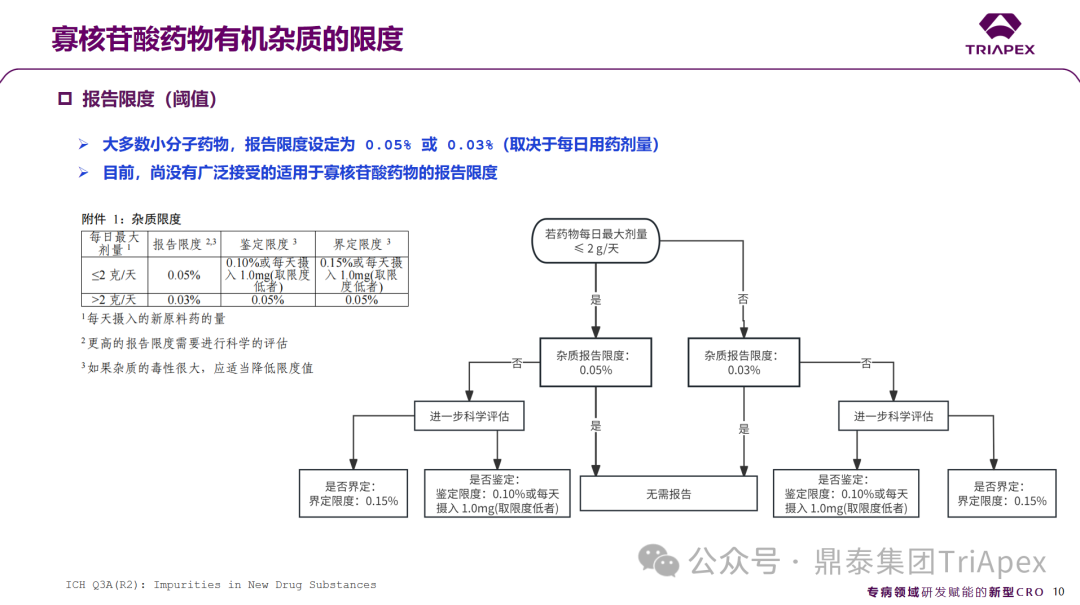


2
对于杂质研究的安全性考量
对于杂质安全性角度的考量主要集中在对杂质界定的定义上。对于小分子化药,杂质界定被定义为“获得和评价某些数据的过程,这些数据可用于确保单个杂质或在特定的含量下的一系列杂质的生物安全性”[2]。根据ICH Q3A,对于一个通过充分的非临床安全性研究和临床研究的新原料药,其任一杂质的水平已经得到合理界定。对于动物/人体中的重要代谢物的杂质,通常也视为已通过界定。如果杂质的界定水平(Qualified level)高于告诉实际所含的杂质量(质量标准限度,Specification limit),同样可以根据完成的安全性研究中使用的实际杂质量来判断其合理性(图1)。
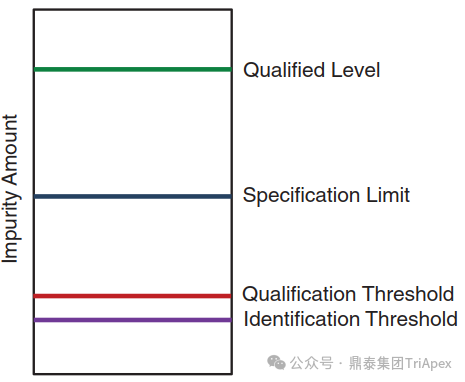
图1. 安全性数据(Qualified Level)、质量标准限度(Specification Limit)、鉴定限度(Identification Threshold)和界定限度(Qualification Threshold)的关系[1]
如果无法获得足够数据支持拟定的限度且超过界定限度,则必须开展进一步研究以支持该限度的合理性。对于某些药物,可以根据科学原理和认知水平(如药物的类别和临床经验),对其杂质界定限度进行适当调整。总之,对限度的确定应具体情况具体分析。上述基本原则同样适用于siRNA 药物。
siRNA 药物杂质界定研究策略
1
识别杂质类别
根据结构,可将杂质分为四类,每类杂质的安全评估要求不同:
Class I:与siRNA药物序列相同的代谢产物类杂质,如缺少末端碱基的杂质,其安全性已在非临床评价中确立,不需要进一步评估。
Class II:仅含天然核酸中存在的结构元素的杂质,如磷酸二酯杂质,可以视为安全,不需要进一步评估。
Class III:与siRNA药物序列不同但不含新结构元素的杂质,如n-1、n+1,单个杂质水平很难达到危害水平,一般不需要动物试验。
Class IV:含siRNA药物和天然核酸都不具有的新结构元素的杂质,若其限量超过1.5%的界定限度时,需进行非临床安全性研究。
2
非临床评价设计
为了对Class IV类杂质进行界定而进行非临床研究时,动物种属、供试品、研究周期和剂量水平是重要的设计要素。

动物种属:与ICH Q3A中给出的建议一致,该研究可仅在一种啮齿类动物中进行。
供试品:尽管选择生产仅含有杂质或杂质混合物的样品是可以接受的,通常更倾向于使用由掺入了相关杂质的siRNA药物,主要是因为:1)生产富含杂质的siRNA药物往往比生产单纯的杂质更实用;2)在siRNA 药物存在的情况下,评估杂质的安全性更能代表杂质在患者中暴露的真实情况。
许多常见siRNA 药物杂质,例如 abasic 和 CNET 杂质,均为单个成分组成的混合物,它们之间的区别仅在于序列中的位置。对于该类杂质,评估其中的一个代表性杂质通常足够,但评估同一类杂质的混合物也是允许的,而且由于合成方面的考虑,有时实际上也是不可避免的。根据经验,最有效的杂质评估方法是采用盒式设计(Cassette maner),将siRNA 药物与一种以上的杂质配比混合后共同给药。
研究周期:一般来说,研究周期应足以确保在值得关注的组织中达到稳态浓度。对半衰期较长、计划长期使用的siRNA,可能需要长达13周的给药(表1);对于在组织中半衰期较短或不计划长期使用的siRNA,可以短期给药。给予的杂质的量应等于或超过期望的覆盖水平。
剂量水平:建议至少设置2个剂量水平,低剂量应为预期的NOAEL,高剂量应扩大治疗窗口或足以产生与siRNA 药物一致的毒理学效应。除了掺杂的供试品组外,还应设置给予供试品或溶媒的对照组。每组动物数应足够,如10只/性别/组。
表1. 小鼠重复给药13周杂质界定试验设计[1]
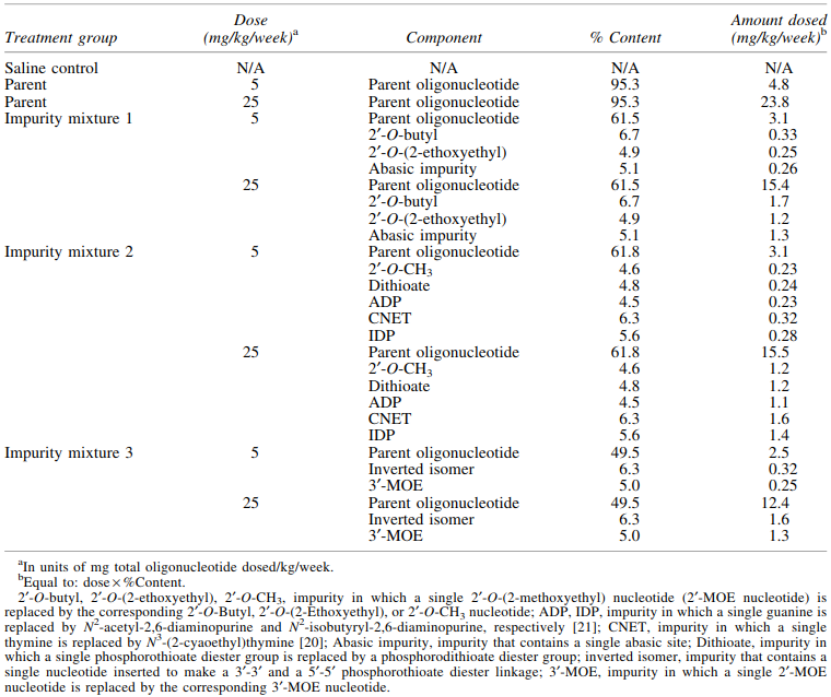
3
毒理学评估
毒理学评估的指标应包括:in-life 观察(如体重、摄食量、临床观察和眼科检查)、临床病理(包括血液学和血清生化等)、大体解剖、器官重量和组织病理学检查;还应检测siRNA在组织中的浓度,以评估暴露情况。

4
计算毒理学覆盖范围
在研究中产生的毒理数据可以用来计算,主要有两种方法。一种简单的方法是将供试品中杂质百分比设为覆盖率,如安全剂量大于或等于临床剂量,该方法是有效的;另一种更为灵活的方法是计算适当剂量下供试品中实际存在的杂质量,以相对于临床剂量的百分比表示,作为覆盖范围(表2)。
表2.计算毒理学覆盖范围的可能方法[1]
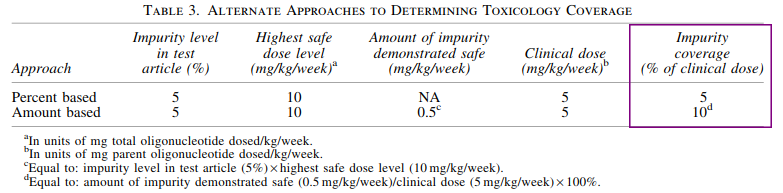
5
siRNA药物杂质界定决策树
对于小分子化学药而言,降低杂质含量使其低于限度通常要比提供杂质的安全性数据来得简单。根据ICH Q3A中的推荐的决策树,当杂质含量超过限度且已有信息不足以提示其安全性时,应考虑进行额外的安全性试验。如何合理地界定一个杂质将取决于许多因素,包括患病人群、每日剂量、给药途径与疗程。
对于siRNA药物来说,通过工艺优化进行杂质水平的控制可能同样是适用的,只不过限于固相合成工艺的特殊性和生产成本方面的考虑可能需要综合研判。对于siRNA药物杂质的界定决策树,有其自身的特殊之处,主要体现在触发杂质界定研究的条件和逻辑不完全相同、需要开展的界定试验也不相同。
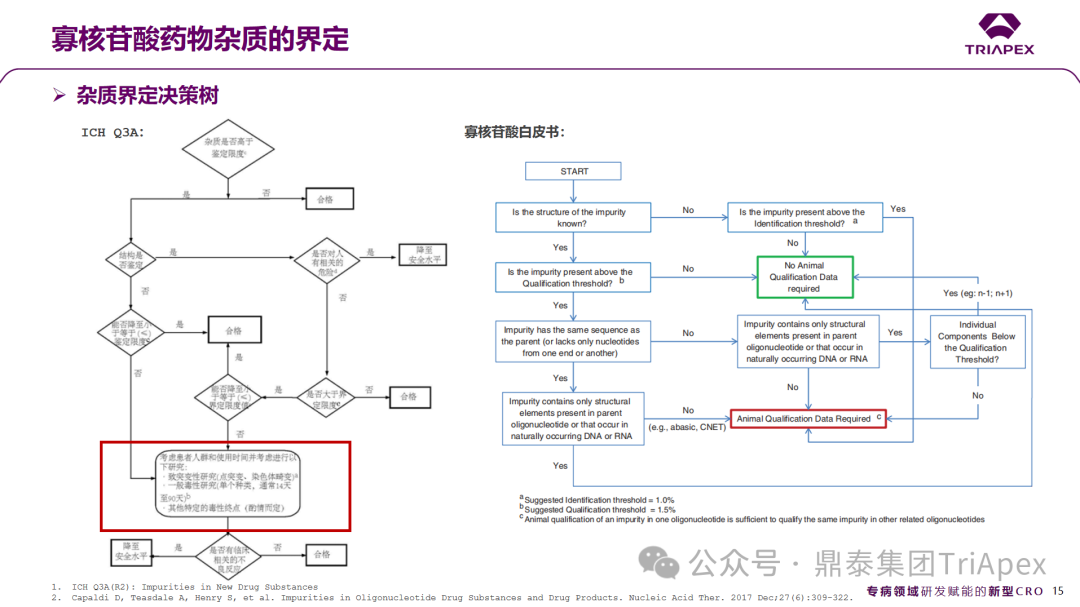

已上市 siRNA 药物杂质控制案例分析





结语
大部分siRNA药物相关杂质主要包括与起始物料、生产工艺等相关的杂质,建议对杂质进行归类和报告。鉴于检测方法的限制,siRNA 药物杂质的报告限度、鉴定限度和界定限度采用高于ICH Q3A中的相应水平可能是合理的。对于含有在siRNA原形药物或天然核酸中所没有的结构元素的杂质,需要进行非临床安全性评价。在啮齿类动物重复给药毒性试验中,可伴随进行杂质安全性评估。毒理批次的杂质谱宜涵盖临床批次的杂质谱。
本文综合相关指南、文献资料及FDA 已批准上市siRNA 药物的案例分析,就siRNA药物杂质问题提供了一定的科学建议,旨在确保药物的质量和安全性,并指导非临床研究设计和助力临床转化。
参考资料:
[1] Daniel Capaldi, Andy Teasdale, Scott Henry, et al. Impurities in Oligonucleotide Drug Substances and Drug Product. NUCLEIC ACID THERAPEUTICS. 2017
[2] ICH Q3A(R2). Impurities in New Drug Substances. https://www.ich.org/page/quality-guidelines







 法规政策
法规政策 项目管理
项目管理


 Global
Global



 苏公网安备32011202001081
苏公网安备32011202001081