



在前几期文章中,鼎泰团队先后对 3 款已上市 siRNA 药物——Patisiran(Onpattro®,点击查看原文)、Givosiran(Givlaari®,点击查看原文)和 Lumasiran(Oxlumo®,点击查看原文)的上市历程、临床开发路径以及如何从非临床向临床转化进行了较为全面、详细的复盘与总结,为 siRNA 药物非临床研究的设计、结果解读,临床开发路径的规划以及支持产品快速上市的研发和药政策略制定提供了参考和启发。 与前 3 款 siRNA 药物相比,Inclisiran 的特别之处在于它是第一款和目前唯一一款获得 FDA 批准用于慢性疾病治疗的 siRNA 药物,适应症为: 作为饮食的辅助疗法,用于治疗杂合子家族性高胆固醇血症(HeFH)或临床动脉粥样硬化性心血管疾病(ASCVD)的成人患者,这些患者接受了最大耐受剂量的他汀类药物治疗,但需要额外降低低密度脂蛋白胆固醇(LDL-C)。 LEQVIO® is indicated as an adjunct to diet and maximally tolerated statin therapy for the treatment of adults with heterozygous familial hypercholesterolemia (HeFH) or clinical atherosclerotic cardiovascular disease (ASCVD), who require additional lowering of low-density lipoprotein cholesterol (LDL-C). Inclisiran 从递交 IND 申请到首个适应症获批上市历时约 6 年,随后陆续获 EMA、NMPA 及 PMDA 批准上市。 Inclisiran 的获批上市掀开了 siRNA 药物用于慢病治疗的新篇章,将引领更多 siRNA 候选分子用于代谢性疾病、感染性疾病和心血管疾病等领域。 目前,国内已有 6 款以 PCSK9 为靶点的 siRNA 药物进入临床试验,后续也还将陆续有更多优秀的同类分子进入临床。截至目前,鼎泰团队已完成了 60 多 个 siRNA 药物的相关研究,其中以 PCSK9 为代表的拟用于代谢或心血管疾病的 siRNA 近 10 个,积累了丰富的评价经验,包括非人灵长类或转基因小鼠药效学、药代动力学和毒理学研究。 Inclisiran 是全球首款获批用于慢性疾病治疗的 siRNA 药物,也是我国截至目前批准的唯一一款 siRNA 药物,同时也是唯一一款同时包括单药和联合用药方案的 siRNA 药物,其临床开发路径、药政策略和相关的支持性非临床研究具有重要的参考价值。基于既往的项目经验和对未来产品开发预期的规划,鼎泰团队对 Inclisiran 上市历程及临床开发路径进行了复盘,学习他山之石、寻找突破创新思路,期待为助力该类药物更高效地实现临床转化和推动产品上市提供借鉴和启示。 FDA 和 NMPA 批准的产品外包装
★ 文章导览 ★ 1 产品的研发背景 关键词:他汀类药物、PCSK9、联合用药 2 Inclisiran 产品信息 3 Inclisiran 临床开发路径 关键词:起始剂量、3 项关键Ⅲ期 4 监管互动里程碑 关键词:EOP2、ADA 检测、Pre-NDA 5 免疫原性检测与药代动力学 6 非临床与临床试验相关性分析 7 小结 产品的研发背景
1、疾病背景 心血管疾病(Cardiovascular Disease,CVD)一直是全球一大死亡原因,ASCVD 是最常见的一种 CVD,而高胆固醇血症是 ASCVD 的诱因之一。特别是以 LDL-C 或总胆固醇(Total Cholesterol,TC)升高为特点的血脂异常是 ASCVD 重要的风险因素。因此,控制 LDL-C 对心脏病和非心脏病患者至关重要[1]。 2、目前治疗手段 ASCVD 的管理包括生活方式的改变,如有氧运动和调节饮食。除了健康的生活方式干预外,还可以使用降脂药物进行药物治疗。 目前,降脂药物主要有他汀类药物、胆固醇吸收抑制剂、贝特类药物、抗氧化类药物(普罗布考)、烟酸类药物、PCSK9 抑制剂(PCSK9:靶向降解编码前蛋白转化酶枯草溶菌素 9 )等。 降脂药物对肝细胞的作用机制[2] ① 他汀类药物 ② PCSK9 抑制剂 LDL-C 是临床血脂检测的四项重要指标之一,其水平越高,患 ASCVD 的风险就越高。PCSK9 是肝脏合成的分泌型丝氨酸蛋白酶。在细胞外,PCSK9 与肝细胞表面的 LDL-R 结合并使其降解,从而阻止 LDL-R 的再循环,导致细胞表面 LDL-R 浓度降低,血浆 LDL particle 浓度增加。 PCSK9 抑制剂可阻断 PCSK9 与膜结合LDL-R(membrane-bound LDL receptor)的相互作用,或直接阻断 PCSK9蛋白的合成(siRNA 类),使参与体循环的 LDL-R 增加,细胞表面受体浓度增加,提高 LDL-R 对 LDL-C 的重摄取,降低循环 LDL-C 水平,最终达到降低血脂的目的(见下图)[4]。研究表明,在降低血清胆固醇水平方面 PCSK9 抑制剂比他汀类药物更有效[6]。 Ezetimibe 和/或 PCSK9 抑制剂被认为是在接受最大耐受剂量他汀类药物治疗的 CVD 高风险(二级预防和 HeFH)、需要额外降低 LDL-C 的患者的二线加载治疗(addon therapy)[3]。 PCSK9 介导 LDL-C 降解的作用机制[4] 3、PCSK9 与他汀类药物联合使用 尽管他汀类药物是最常用的降血脂药物,但其在治疗过程中存在一定的局限性,如 CVD 事件的风险仍存在、LDL-C 水平未达临床治疗目的、部分患者对他汀类药物不耐受和他汀类药物依从性差等[5]。 研究表明,与单独使用他汀类药物相比,他汀类药物 + PCSK9 抑制剂的辅助治疗可以使 LDL-C 水平降低 50-60%。此外,PCSK 抑制剂联合高剂量他汀类药物可降低临床 ASCVD 患者的 CVD 事件和全因死亡率(all-cause mortality)[4]。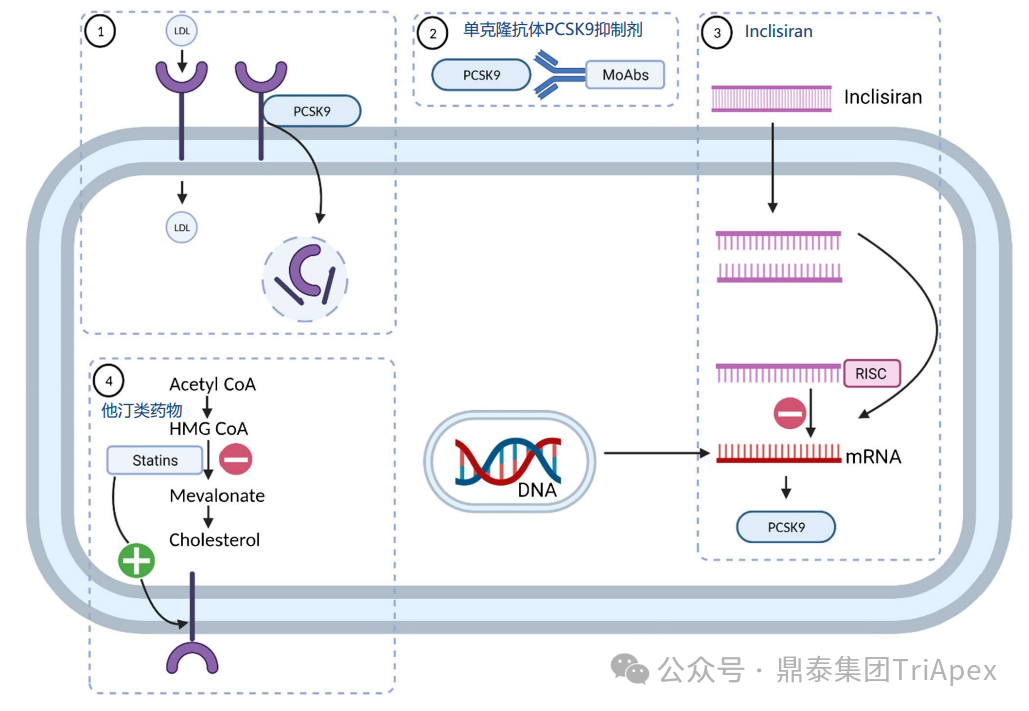
Inclisiran 产品信息
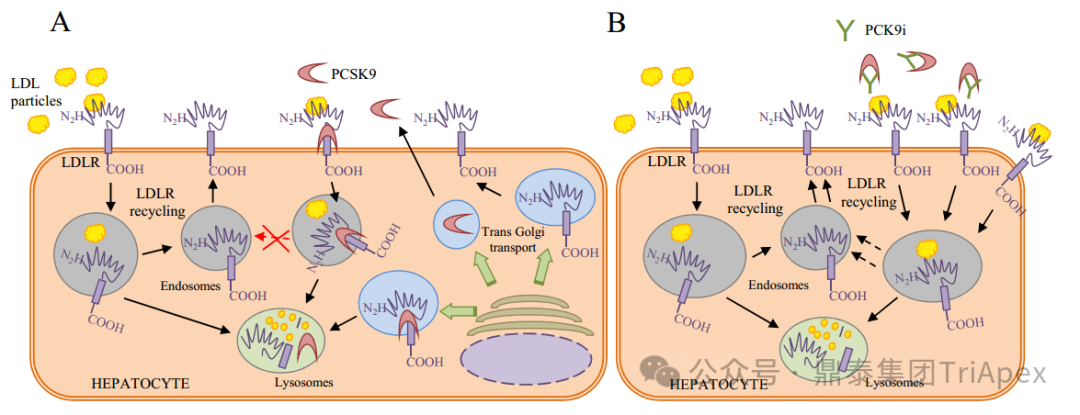
Inclisiran 是一款 GalNAc 偶联的 siRNA 药物(GalNAc-siRNA),靶向肝脏分布。Inclisiran 与单抗类 PCSK9 抑制剂的最大不同点:Inclisiran 可直接作用于编码 PCSK9 蛋白的 mRNA,利用 RNA 干扰机制,诱导 PCSK9 mRNA 降解,增加肝细胞表面 LDL-R 的表达和循环,继而增加对 LDL-C 的摄取,并降低循环中的 LDL-C 水平(见下图)。 PCSK9 蛋白的生物学功能和 Inclisiran 的作用机制示意图[7] Inclisiran 最初由 Alnylam Pharmaceuticals 研发,于2019 年 11 月由诺华(Novartis)收购 The Medicines Company(该公司从 Alnylam Pharmaceuticals 购买了 Inclisiran 分子的授权)后进行后续开发。 2020 年 12 月 09 日,Inclisiran 首先在欧盟获批上市,作为饮食的辅助疗法,用于成人原发性高胆固醇血症(杂合子型家族性和非家族性)或混合型血脂异常患者的治疗[8]。 2021 年 12 月 22 日,Inclisiran 获得 FDA 批准,适应症为:作为饮食的辅助疗法,用于治疗杂合子家族性高胆固醇血症(HeFH)或临床动脉粥样硬化性心血管疾病(ASCVD)的成人患者,这些患者接受了最大耐受剂量的他汀类药物治疗,但需要额外降低低密度脂蛋白胆固醇(LDL-C)。 2023 年 08 月 22 日,Inclisiran 在 NMPA 获批上市,获批的适应症与 EMA 获批的相同。 2023 年 09 月 25 日,Inclisiran 在 PMDA 获批上市,作为饮食的辅助疗法,用于成人原发性高胆固醇血症的治疗。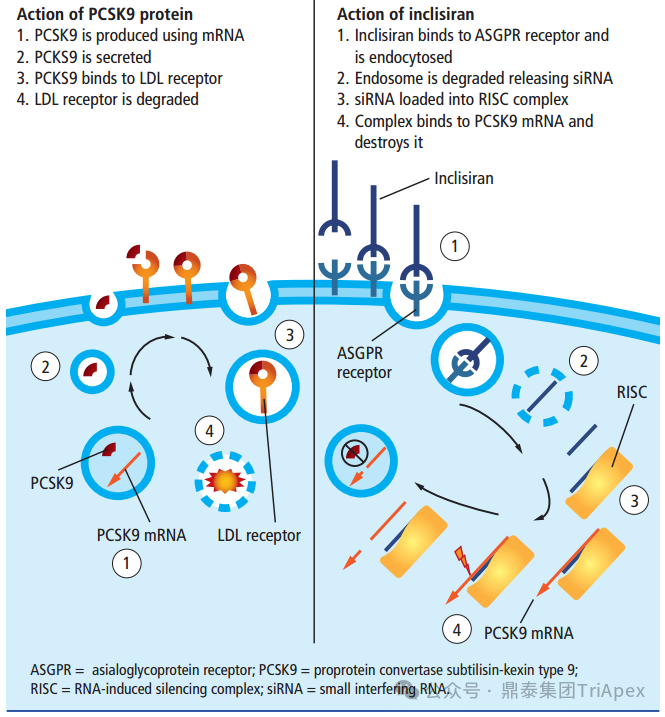
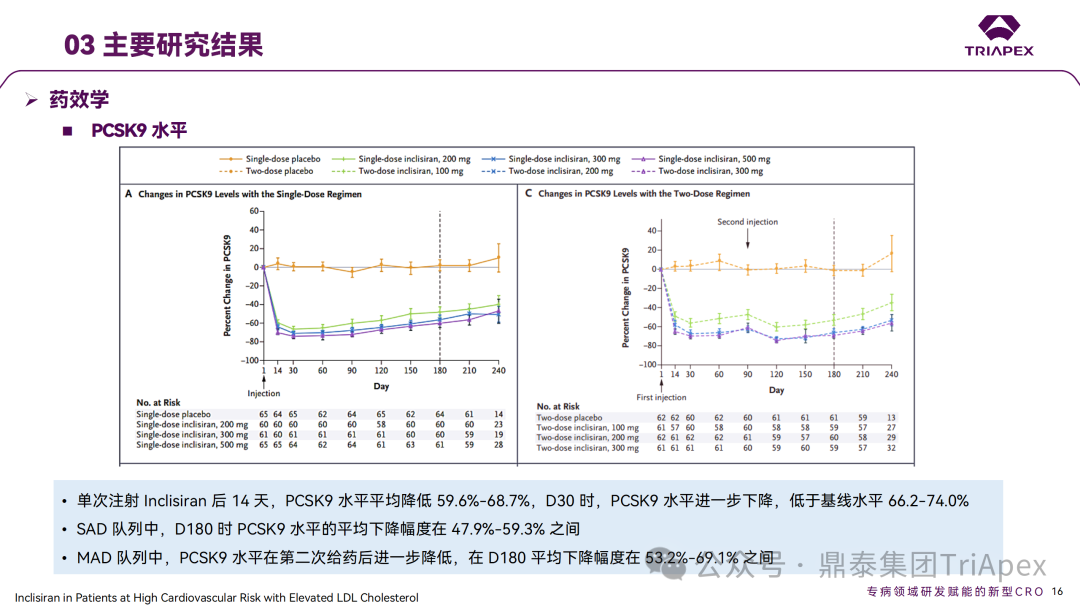
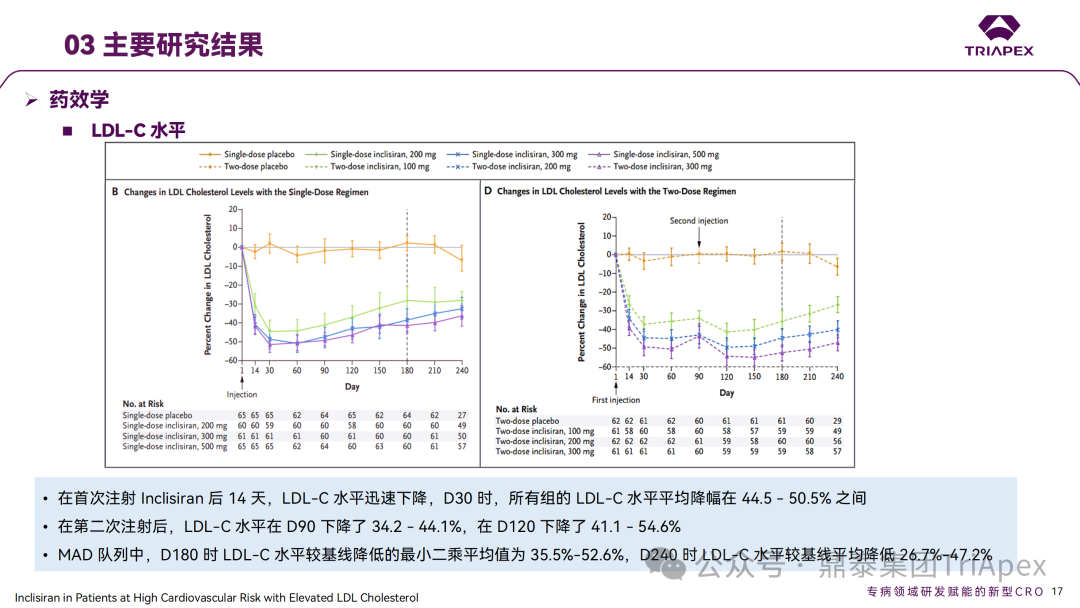
Inclisiran 临床开发路径
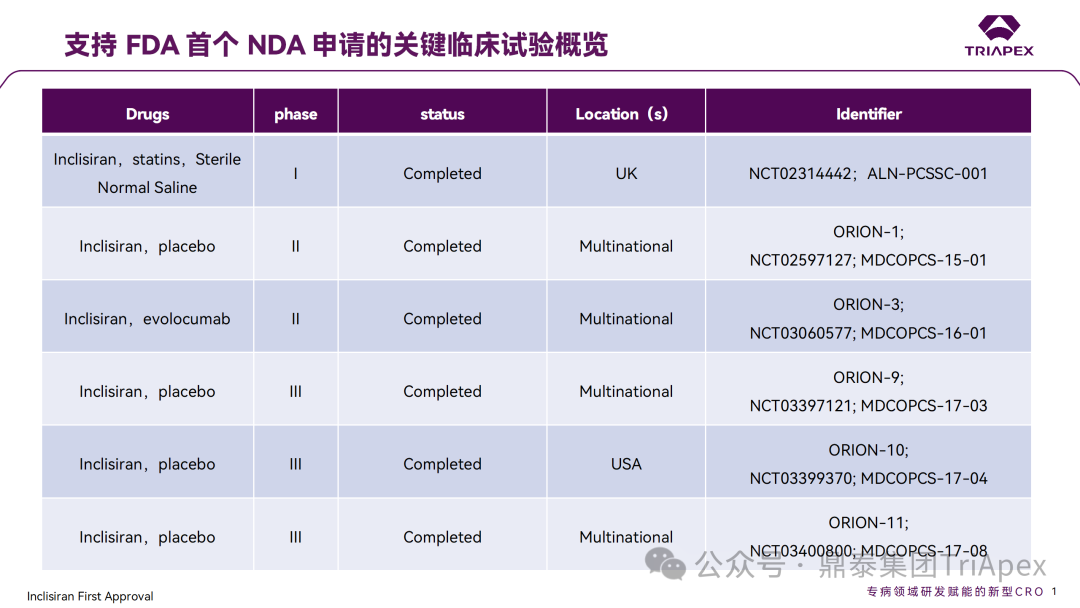
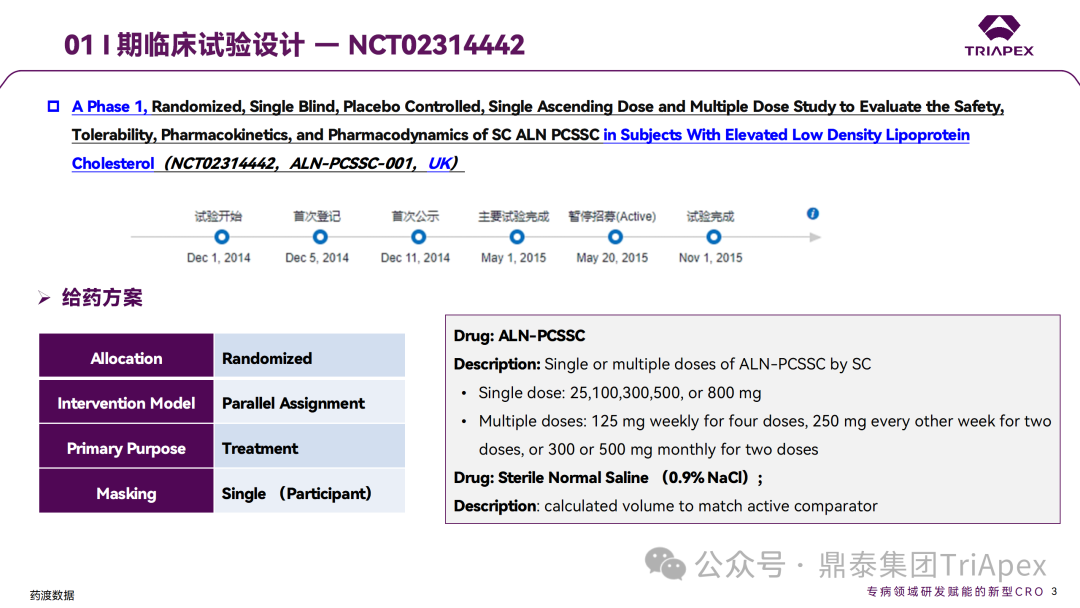
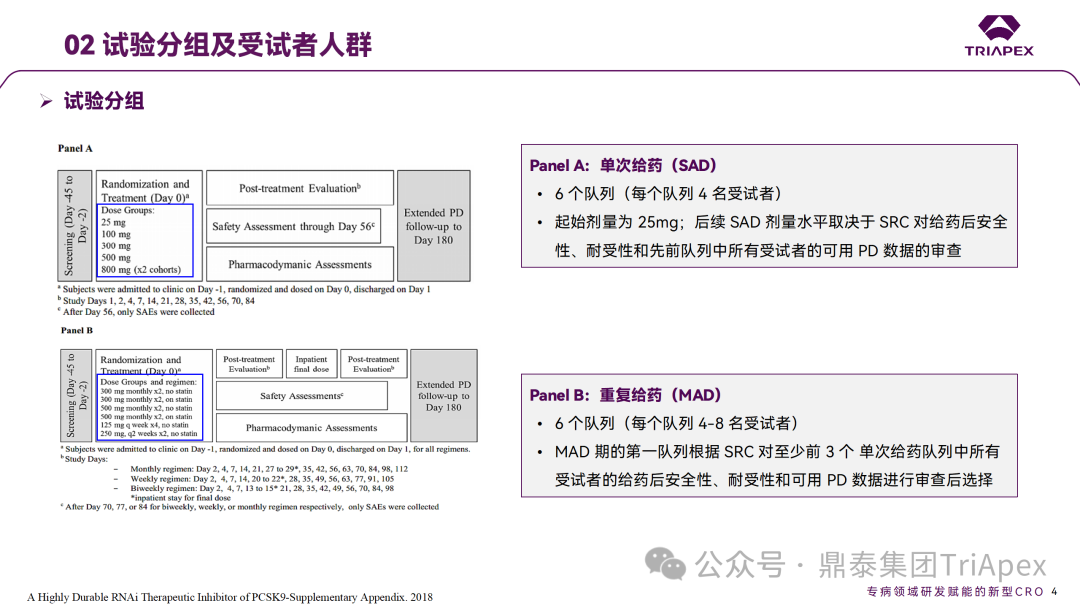
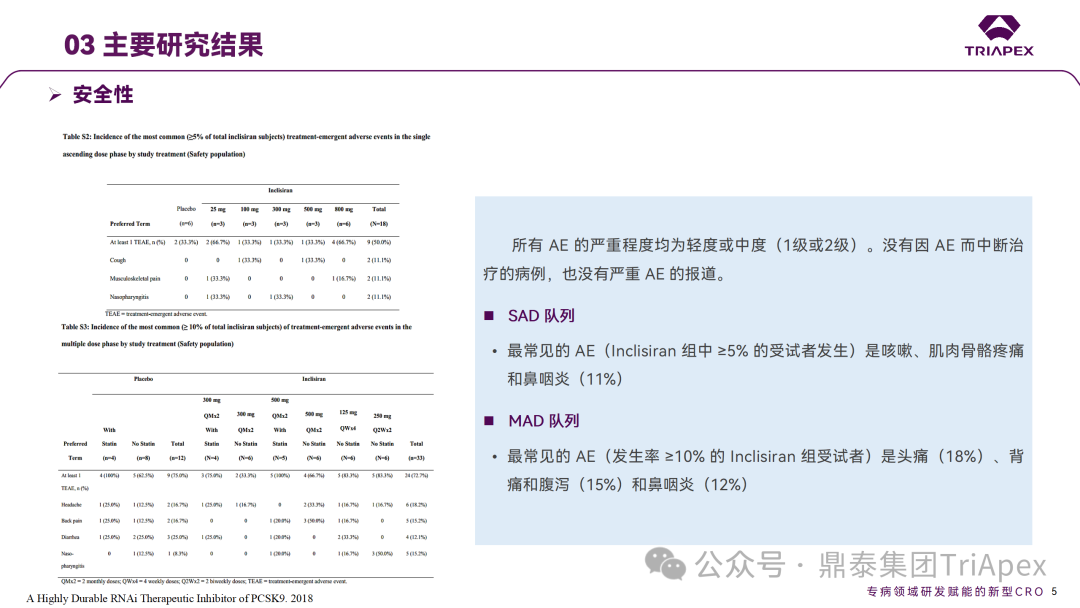
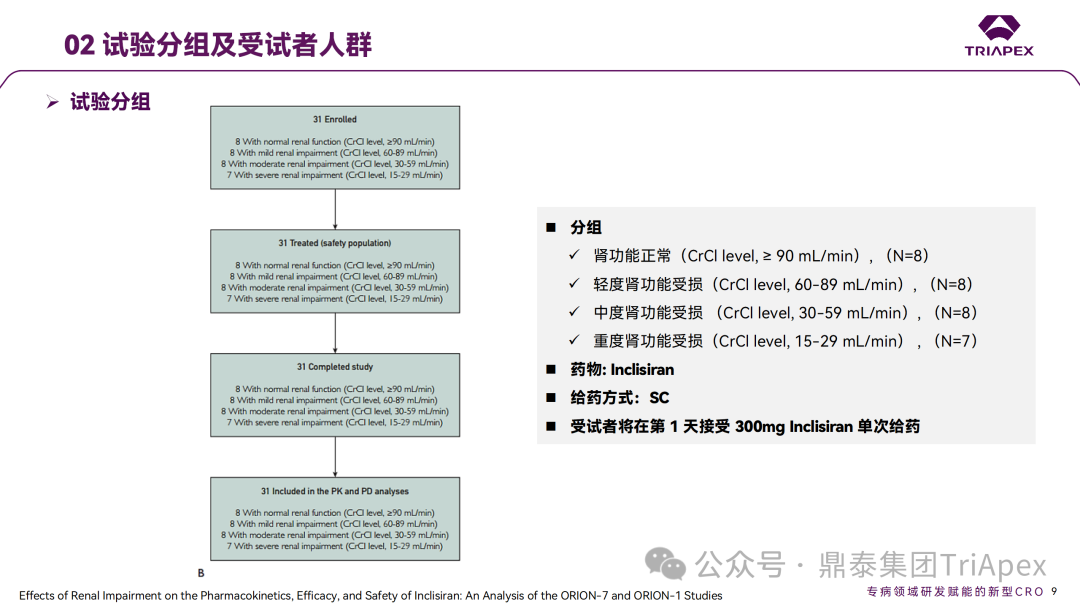
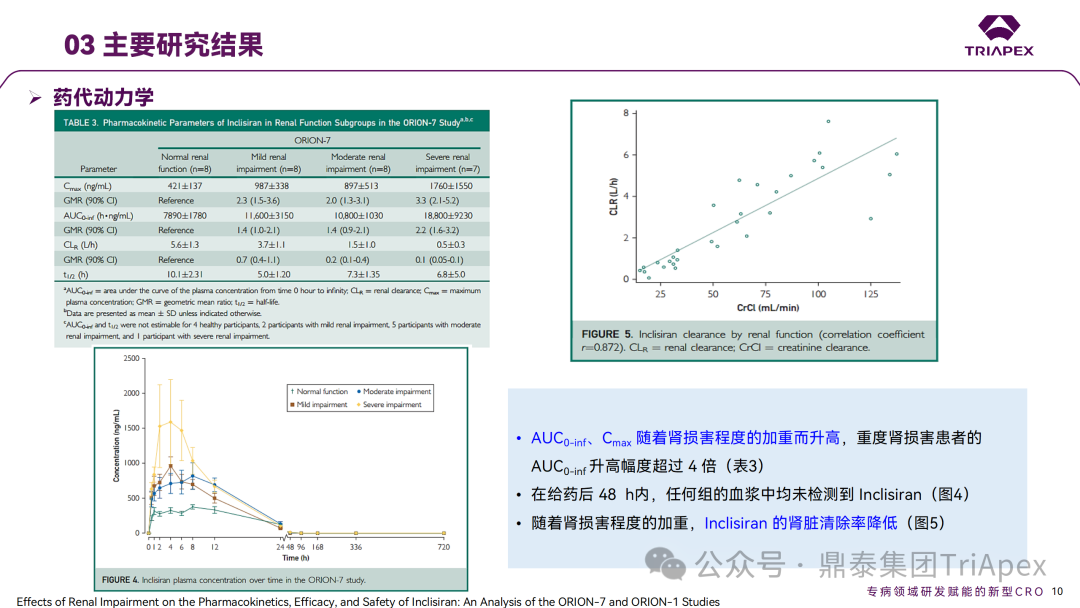
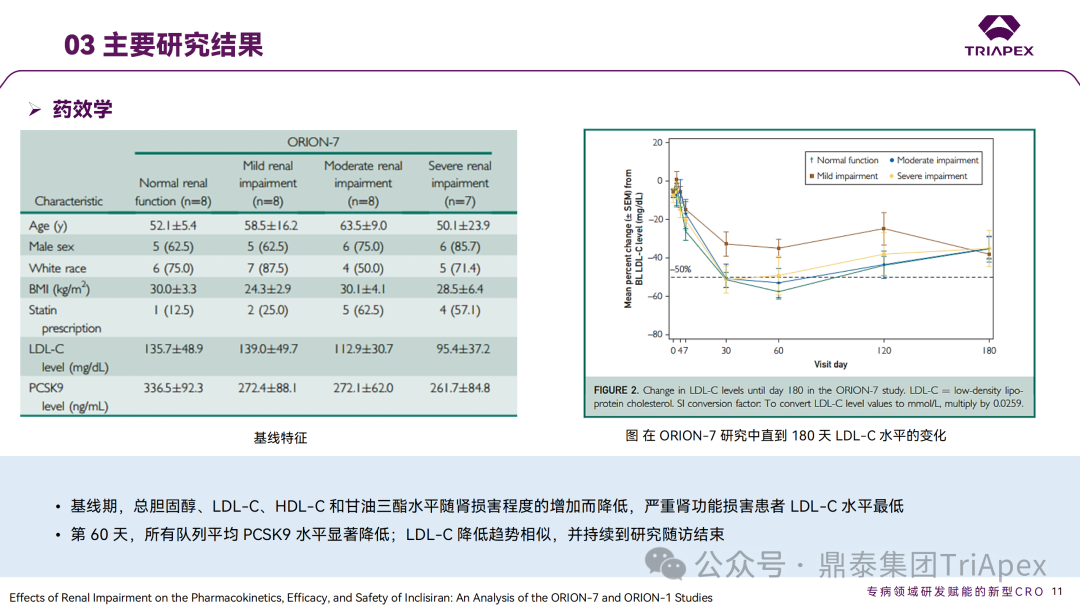
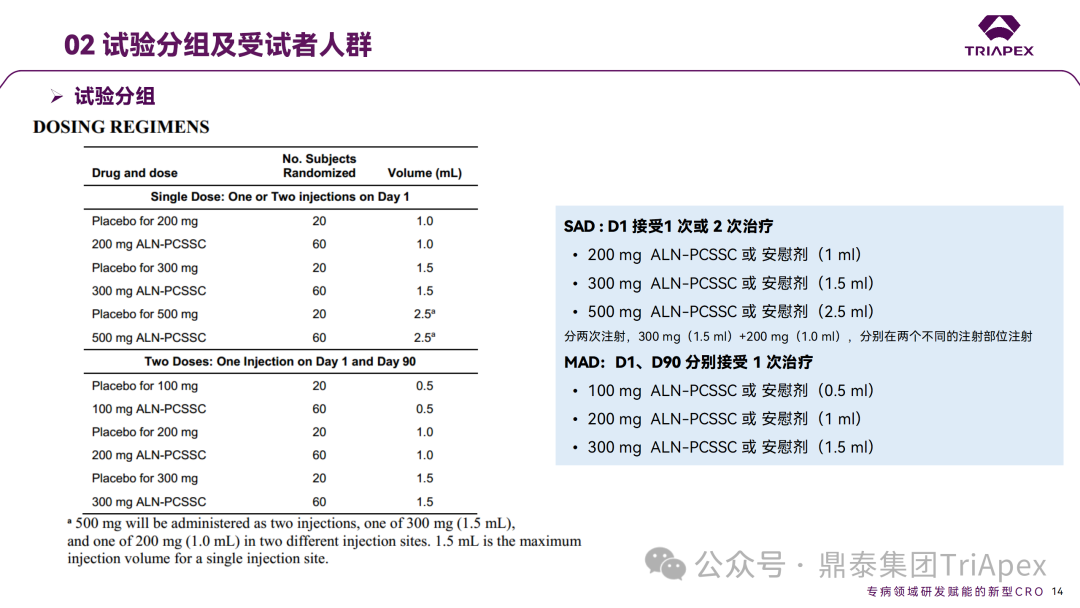
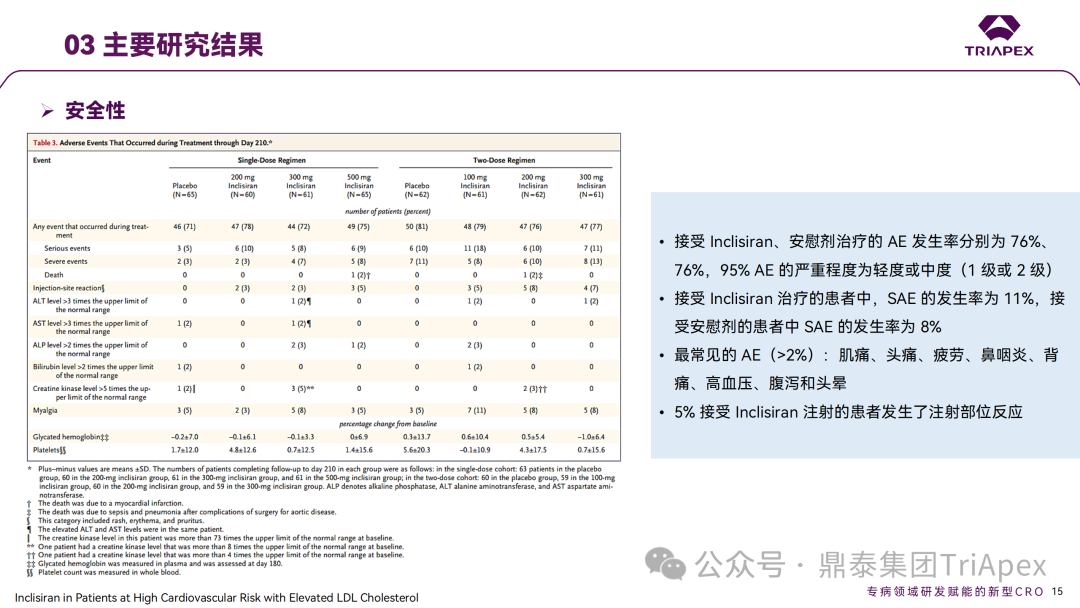
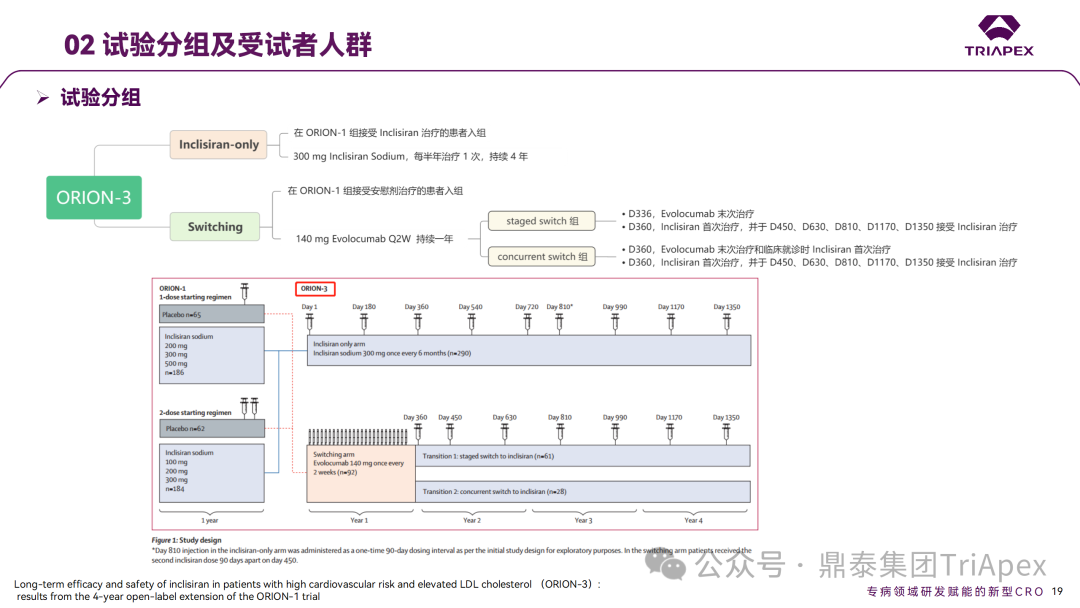
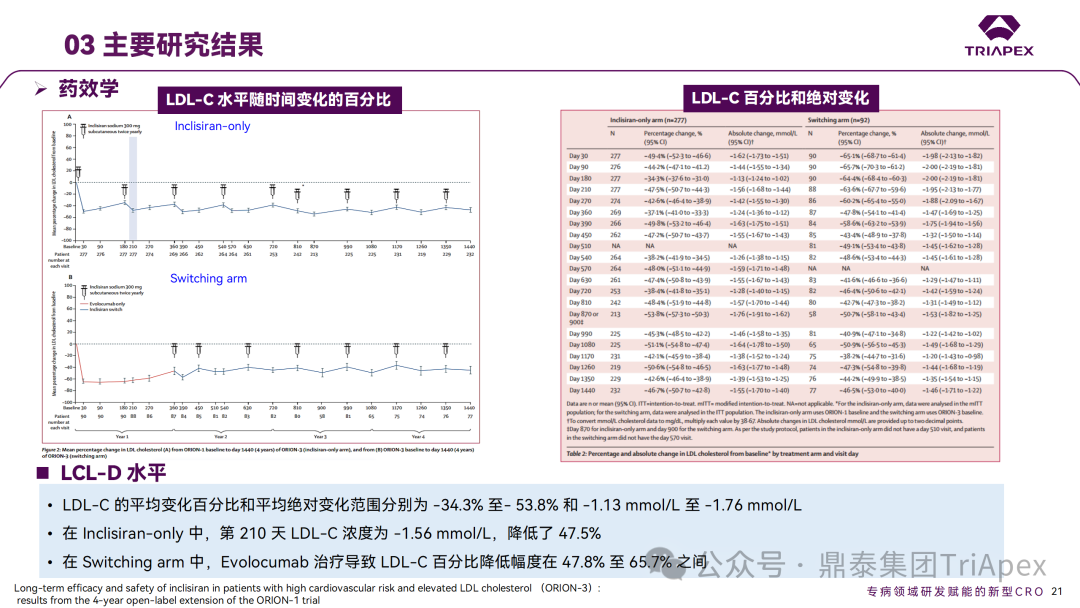
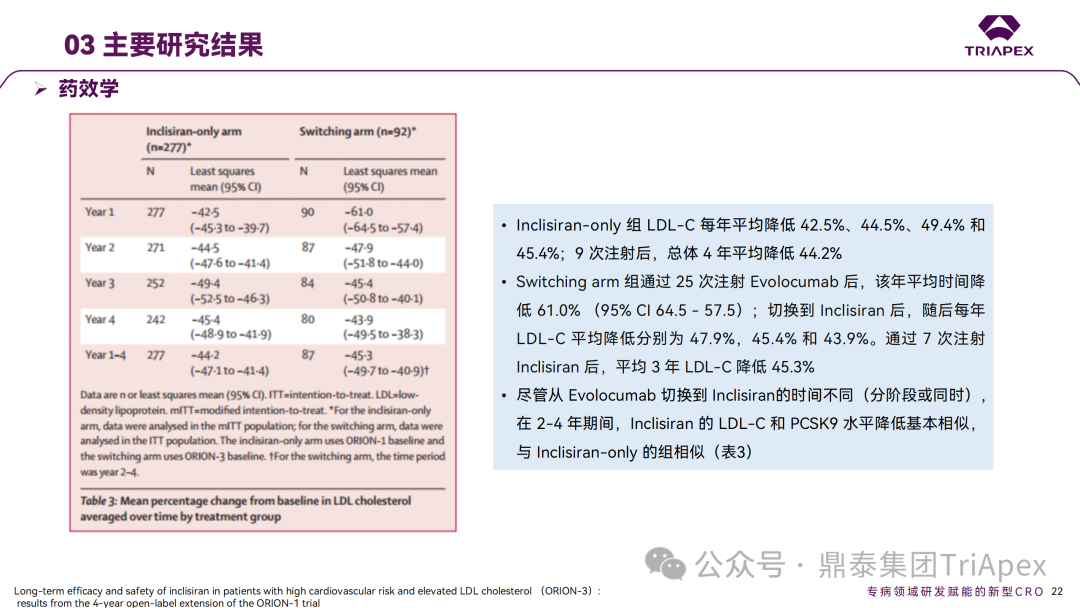
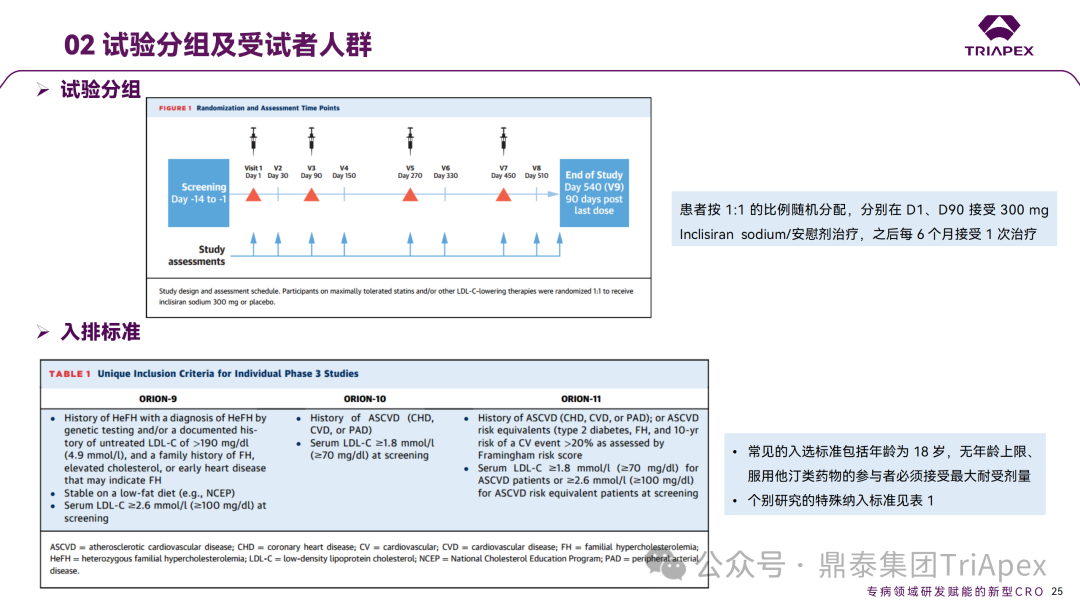
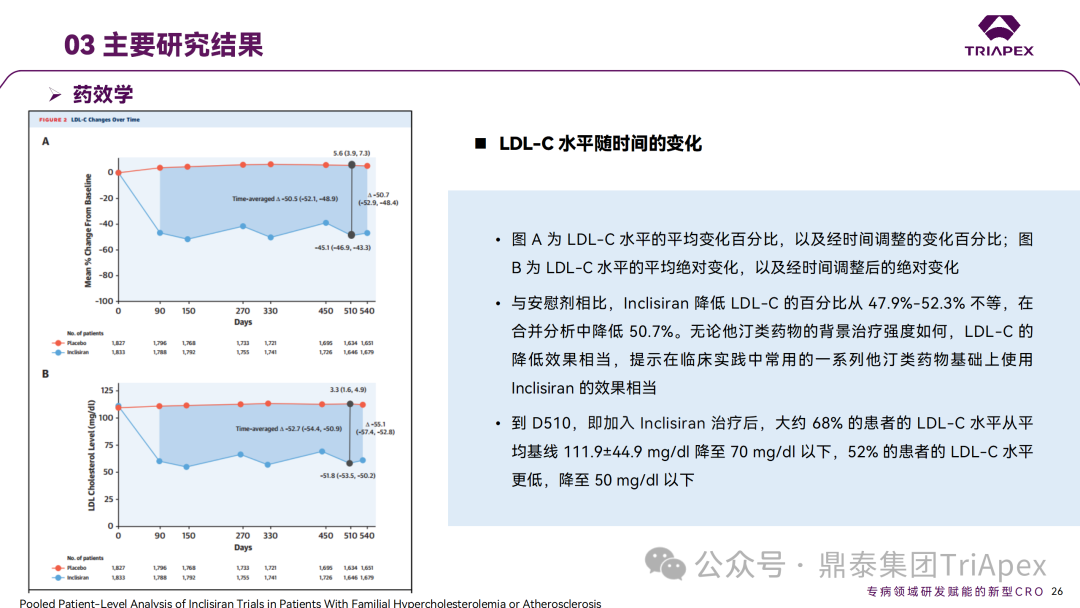
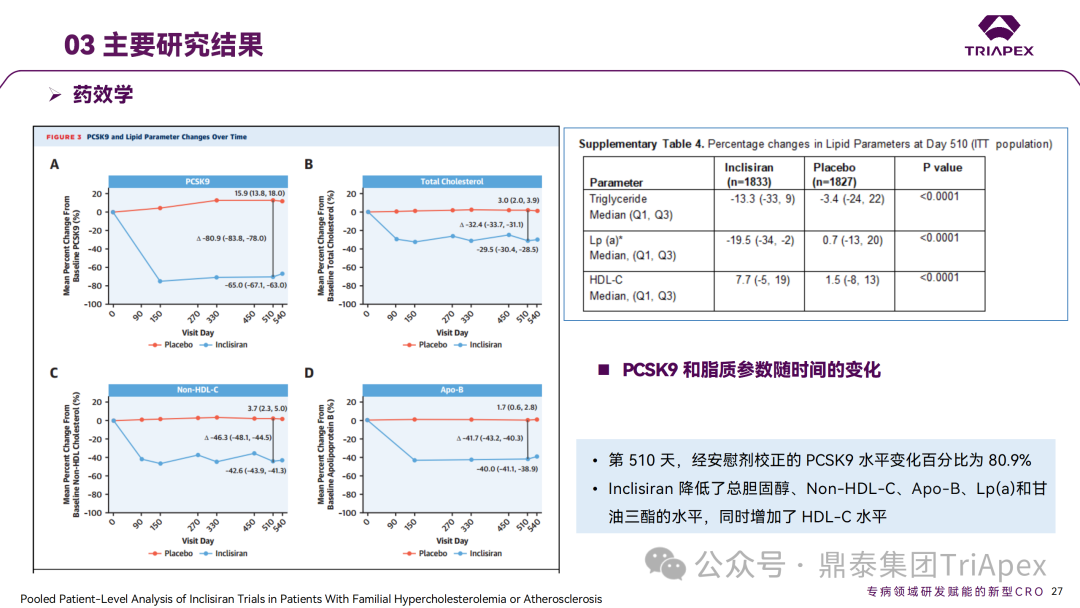
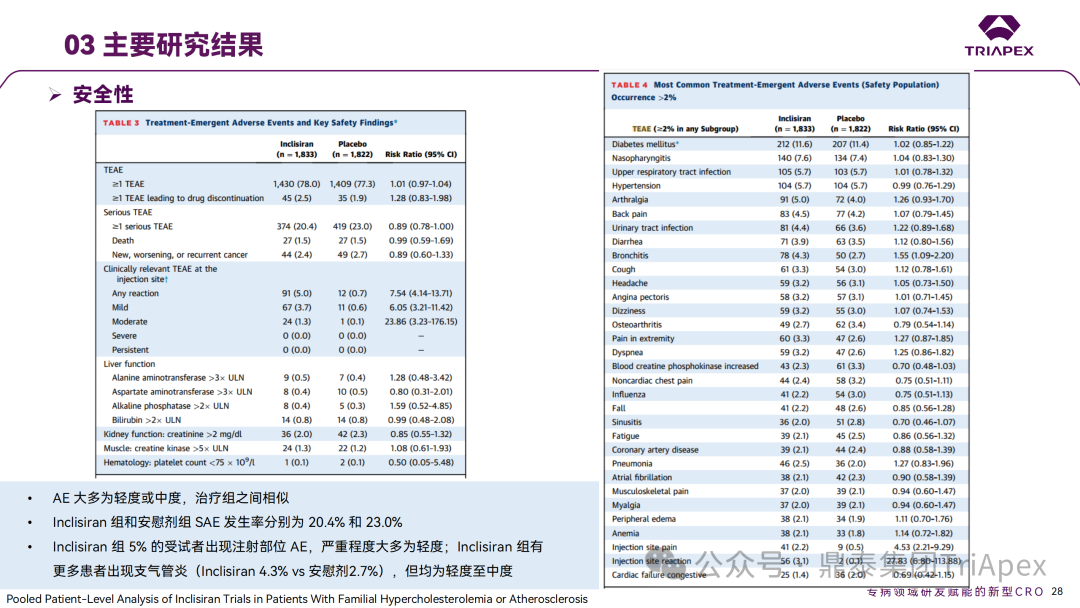
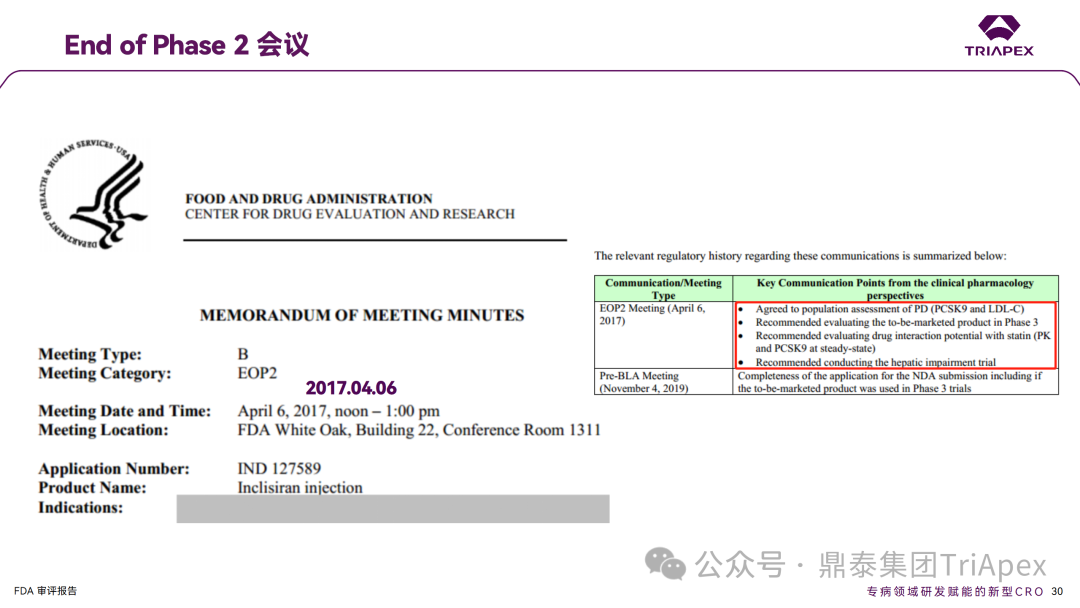
Inclisiran 从 IND 申请到获批上市历时约 6 年时间,支持 Inclisiran 首次 NDA 批准的主要临床试验包含 1 项 Ⅰ 期试验、2 项 Ⅱ 期试验和 3 项 Ⅲ 期临床试验。 临床开发路径小结 支持首个 NDA 申请的临床试验 1、Ⅰ 期临床 Ⅰ 期临床起始剂量设计依据 在大鼠和猴重复给药 28 天毒性试验中,确定了 NOAEL 剂量分别为 250 mg/kg 和 300mg/kg,该结果支持 Ⅰ 期临床试验 FIH 起始剂量为 25mg(具体计算过程未透露)。 Ⅰ 期临床起始剂量设计依据 ① NCT02314442-ALN-PCSSC-001,UK 2014 年,Inclisiran 的首次人体试验(FIH)启动。这是 1 项在英国开展的随机、单盲、安慰剂对照、剂量递增试验,旨在评估 Inclisiran 在 LDL-C 升高受试者中的安全性、耐受性、药代动力学和药效学。 Ⅰ 期临床试验设计 • 125mg QW*4 单药 • 250mg Q2W*2 单药 • 300mg QM*2 单药 • 300mg QM*2 与他汀类药物联合用药 • 500mg QM*2 单药 • 500mg QM*2 与他汀类药物联合用药 主要终点为 AE、SAE 和导致研究药物停药的 AE 的受试者比例;次要终点为药代动力学参数(Cmax、Tmax、AUC、t1/2)及 Inclisiran 对血清 LDL-C 水平、血浆 PCSK9 水平的影响。 Ⅰ 期临床试验分组和给药方案 所有 AE 的严重程度均为轻度或中度;在 1 个月内给予 Inclisiran,观察到 PCSK9 和 LDL-C 水平持续下降;服用稳定剂量他汀类药物的患者在给予 Inclisiran 后可进一步降低 LDL-C 水平。此外,本研究支持 PCSK9 作为显著降低 LDL-C 的治疗靶点。 探索性研究结果显示,无论 SAD 还是 MAD,接受 Inclisiran 治疗的受试者的 TC、非高密度脂蛋白胆固醇(Non-HDL-C)和载脂蛋白 B(Apo-B)水平均有所下降;与安慰剂组相比,从基线到接受首次治疗后第 84 天时,上述指标均显著降低。 Ⅰ 期临床试验安全性相关结果 Ⅰ 期临床试验药效学相关结果 ② NCT03159416-ORION-7,New Zealand 肾清除是 Inclisiran 体内消除的主要途径,在给药后 24h 内尿液中检测到约 1/3 的总给药剂量。ASCVD 患者常伴有肾功能不全,因此,有必要对Inclisiran 在不同程度肾功能损害个体中的安全性、PK 和 PD 效应进行表征。 NCT03159416-ORION-7 是一项在新西兰开展的单次给药、开放标签、平行组研究,以评估肾功能损害与肾功能正常受试者的药代动力学。该试验根据肾功能损害的程度分为四组,分别为肾功能正常组、轻度肾功能损害、中度肾功能损害和重度肾功能损害。受试者在第一天接受 300mg Inclisiran 单次皮下给药。 该研究的主要终点为药代动力学参数,包括 Cmax 、Tmax 、t1/2、AUC0-24、AUC0-48、AUC0-inf、SC 给药后的 CL/F 和 Vd/F、排泄分数、肾清除率(CLr)等;次要终点为 PCSK9、血脂、脂蛋白与基线相比的变化。 Ⅰ 期临床试验设计 Ⅰ 期临床试验分组 研究结果显示,在肾功能正常和损害受试者中,Inclisiran 的药效学作用和安全性相似。肾损害患者不需要调整 Inclisiran 剂量。虽然接受 Inclisiran 治疗的肾功能损害患者的血浆暴露量更大,但对 LDL-C 水平、PCSK9 水平的降低程度和持续时间未见影响。该项研究提示了 Inclisiran 可以安全地用于轻度、中度或重度肾功能损害患者,且无需调整剂量或给药方案。 Ⅰ 期临床试验药代动力学结果 Ⅰ 期临床试验药效学结果 除此之外,还开展了一项 Ⅰ 期临床试验—ORION-6,这是一项单剂量、开放标签、平行组研究,以评估肝功能损害受试者与肝功能正常受试者的药代动力学、药效学和安全性。研究结果表明,Inclisiran 可以安全地用于轻度、中度肝功能损害患者,而无需调整剂量或给药方案。 2、Ⅱ 期临床 Ⅱ 期临床剂量设计依据 Ⅱ 期临床剂量是根据 Ⅰ 期研究 ALN-PCSSC-001 的中期结果设计,并将为 Ⅲ 期临床剂量设计提供选择。 Ⅱ 期临床试验剂量设计依据 ① NCT02597127-ORION-1,Multinational ORION-1 是一项 Ⅱ 期、随机、双盲、安慰剂对照试验,共纳入 480 名 ASCVD 或 ASCVD 风险相关患者(ASCVD-risk equivalents,eg, diabetes and familial hypercholesterolemia),这些患者尽管接受了最大耐受剂量的降 LDL-C 治疗,但 LDL-C 水平仍升高。通过单次或多次皮下注射不同剂量的 Inclisiran,评估 ALN-PCSSC 在患者中的有效性、安全性和耐受性。 将患者随机分配至单次给药组或重复给药组: 200mg、300mg 或 500mg Inclisiran 或安慰剂(500mg 分两次注射:300mg+200mg,分别在两个不同的注射部位注射) 100mg、200mg、300mg Inclisiran 或安慰剂 该研究的主要终点是第 180 天 LDL-C 水平相对于基线的变化。安全性随访至第 210 天,LDL-C 和 PCSK9 水平数据可持续检测至第 240 天。 Ⅱ 期临床试验设计 Ⅱ 期临床试验分组 研究结果显示,接受 Inclisiran 治疗的患者 PCSK9 和 LDL-C 水平呈剂量依赖性降低;MAD 队列中,300mg Inclisiran 对 LDL-C 的影响程度最大,48% 患者的 LDL-C 水平在 D180 时<1.3 mmol/L。76% 的受试者出现 AE,大多为轻至中度,最常见的 AE 为肌痛、头痛、疲劳、鼻咽炎、背痛、高血压、腹泻和头晕。该项试验表明,Inclisiran 可降低 LDL-C 水平升高的心血管高危患者的 PCSK9、LDL-C 水平,为后续 Ⅱ 期临床 ORION-7、Ⅲ 期临床试验的剂量选择提供依据。 Ⅱ 期临床试验安全性结果 Ⅱ 期临床试验药效学相关结果 ② NCT03060577-ORION-3,Multinational(ORION-1 的扩展试验) ORION-3 是一项开放标签、多中心、长期(4年)的扩展(OLE)试验,在完成 ORION-1 Ⅱ 期试验的受试者中进行。该项研究纳入的 ASCVD 患者或 ASCVD 风险相关患者尽管接受了最大耐受剂量的他汀类药物治疗,但 LDL-C 仍然升高。来自 ORION-1 的受试者在完成试验后至少到第 210 天,并且 LDL-C 恢复到基线值的 20% 以内,或者患者完成第 360 天的访问(ORION-1),没有 Inclisiran 或 evolocumab 的禁忌症,则有资格参加 ORION-3。此外,受试者必须满足 ORION-3 的所有纳入和排除标准,并签署知情同意书。 该试验包括 2 个治疗组:第 1 组(Inclisiran-only 组)只使用 300mg Inclisiran,每半年治疗 1 次,持续 4 年;第 2 组(Switching组)接受evolocumab Q2W 治疗 1 年后,接受 300mg Inclisiran 治疗,完成剩余的试验。第 2 组受试者按研究机构编号分为两种过渡模式: • 过渡期 1(分阶段):第 336 天最后一次接受 evolocumab 治疗,第 360 天随访时给予首次 Inclisiran 给药,之后每半年治疗 1 次,持续治疗 3 年; • 过渡期 2(同时):第 336 天在家接受最后一次 evolocumab 治疗,第 360 天随访时接受额外的一剂 evolocumab,并且同时接受首次 Inclisiran 治疗,之后每半年治疗 1 次,持续治疗 3 年。 该项临床试验的主要终点为第 210 天 LDL-C 水平从基线到 ORION-3(Inclisiran组)的变化百分比,次要终点为血清 LDL-C、PCSK9 和其他脂质参数相对于基线的变化百分比。 Ⅱ 期临床试验设计 Ⅱ 期临床试验分组 在 Ⅱ 期 OLE 试验 ORION-3 中,最常见的 SAE 是心绞痛和骨关节炎,最常见的 AE 是鼻咽炎、高血压、尿路感染、流感和关节痛。每年两次 Inclisiran 皮下给药耐受性良好,4 年间 LDL-C 和 PCSK9 水平持续有效降低。 ORION-3 表明,Inclisiran 的长期药效学作用不仅具有良好的耐受性,而且通过每年两次的给药,使 LDL-C 的持续额外降低成为可能。 Ⅱ 期临床试验药效学相关结果 3、Ⅲ 期临床 Ⅲ 期临床剂量设计依据 在整个 Ⅲ 期研究期间, Inclisiran sodium 的给药剂量为 300mg。该剂量的设计依据包括:非临床研究中的总体安全性数据、ALNPCSSC-001 Ⅰ 期和 ORION-1 Ⅱ 期研究的临床数据以及多项 PCSK9 抗体研究、群体 PD 模型的数据。 Ⅲ 期临床试验剂量设计依据 ① NCT03397121、NCT03399370、NCT03400800 基于 ORION-9、ORION-10 和 ORION-11 3 项关键的临床试验结果,Inclisiran 的首个 NDA 最终获得 FDA 的批准。这 3 项试验纳入了大约 3660 名 HeFH 或 ASCVD 患者,这些患者尽管接受了最大耐受剂量的他汀类药物治疗,但 LDL-C 仍升高,他们在第 1 天、第 90 天、后续每 6 个月皮下注射 Inclisiran sodium 300mg,持续 18 个月。试验人群包括相关的并发症,如年龄老化、肾功能损害、肝功能损害、高血压和糖尿病等。主要研究终点为 LDL-C 从基线到第 510 天的变化百分比。 Ⅲ 期临床试验 Ⅲ 期临床试验分组 ORION-9、ORION-10 和 ORION-11 3项临床试验的汇总分析显示,Inclisiran 治疗 17 个月后,平均 LDL-C 降低了 50.7%,平均 PCSK9 降低了80.9%;在安全性方面,Inclisiran 组与安慰剂相似,支气管炎和注射部位出现适度的自限性(Modest excess of self-limited)、轻度至中度的 AE;组间肝脏、肌肉、血液学参数无差异。 Ⅲ 期临床试验药效学结果 Ⅲ 期临床试验安全性结果 监管互动里程碑 Inclisiran 的成功上市,与合理的临床试验设计息息相关,当然也离不开与监管的高效互动。在 Inclisiran 的上市历程中,就非临床研究、临床试验设计等问题,多次与监管紧密互动,如 Ⅱ 期临床结束后的 EOP2 会议、上市前的 Pre-NDA 会议等。 监管互动里程碑 1、EOP2 会议 EOP2 会议 在 2017 年 4 月 6 日召开的 EOP2 会议上,申请人就 Inclisiran 的非临床和临床开发计划、临床剂量和给药方案与 FDA 进行沟通,部分临床相关沟通内容如下: Q:FDA 是否同意通过已验证的抗药物抗体(ADA)检测方法进行免疫原性检测的计划? FDA 指出,确定 ADA 是否与内源性治疗药物有交叉反应可以更好地了解治疗风险。由于在目前临床研究中没有检测到 ADA,FDA 没有要求且认为不需要开发这种检测方法。然而,一旦在临床中检测到 ADA,则可能需要额外的检测来充分评估产品的安全性。FDA 建议在第 30 天新增 ADA 的检测,以评估早期 IgG 反应。此外,FDA 还建议申请人在关键临床试验期间收集样品用于 ADA 检测。 申请人的观点是,在 Ⅰ 期或 Ⅱ 期研究中未观察到 ADA,并针对 FDA 建议进行进一步澄清确认(seeking some clarity regarding the request),以确定这些抗体是否与人类 RNA 会产生交叉反应。但由于 RNA 的不稳定性,很难开发出这样的检测方法。因此,申请人认为在观察到 ADA 的情况下检测免疫复合物已足够。 Q:FDA 是否同意拟开展的 PD 参数总体评估方法(没有具体的 PK 参数评估)? 基于 Inclisiran 相对较短的半衰期(约为 7 h)和较长的 PD 效应(单次注射后对 PCSK9 和 LDL-C 的影响,在大多数受试者中持续超过 6 个月),传统的群体 PK 模型不太可能提供关于特殊人群剂量变化的信息,申请人计划从 Ⅲ 期研究的受试者中收集 PD 数据(不进行 PK 采集)来评估 PD 效应的可能差异以及在特殊人群中调整剂量的必要性。 FDA 承认 Inclisiran 的 PK 和 PD 之间存在明显的不相关性,临床药理学信息可使用 PD 数据进行评估,但要有充分的理由和足够的数据来支持 PCSK9 的检测,以评估 Inclisiran 的疗效和安全性。FDA 建议提供 PD 参数总体评估的细节,重点是如何对这些参数和参数的主要应用进行评价。 Q:FDA 是否同意整体的临床药理学方案,并同意他们可以支持最终的 NDA 提交和批准? 申请人正在进行一项体外转运体筛选研究,申请人将根据该项结果及前期研究评估是否需要额外的 DDI 研究。此外,还将对肾和肝功能损害患者进行 Inclisiran PK/PD 研究,以评估是否需要针对上述人群进行剂量调整。 考虑到他汀类药物是拟开展的适应症中重要的联合用药对象,FDA 建议评估包括 PD 在内的 DDI 的可能性。典型的单次给药 DDI 研究可能无法为 Inclisiran 评估 PK 和 PD 相互作用提供足够的数据。因此,FDA 建议在稳定状态下进行研究,例如使用稀疏采样的总体分析方法。或者,将 PCSK9 数据(含或不含他汀类药物)与他汀类药物 PK 一起收集,有助于和使用已有数据(例如 Ⅱ 期研究结果)来评估 PD 相互作用的潜力。 此外,FDA 还建议在 Ⅲ 期临床试验中使用拟最终上市的产品,并在 Ⅲ 期试验之前进行肝功能损害(hepatic impairment)研究。 EOP2 会议临床相关内容问答 申请人在 EOP2 会议中表明会根据研究结果评估 DDI 开展的必要性,但最终并未进行体内 DDI 研究。这是因为 Inclisiran 的成分不是 CYP 450 或转运体的底物、抑制剂或诱导剂。由于临床上有联合用药计划,在毒理学研究中进行了联合用药的毒性评估,未发现对毒性或TK影响[9]。 Inclisiran 与他汀类药物相互作用研究








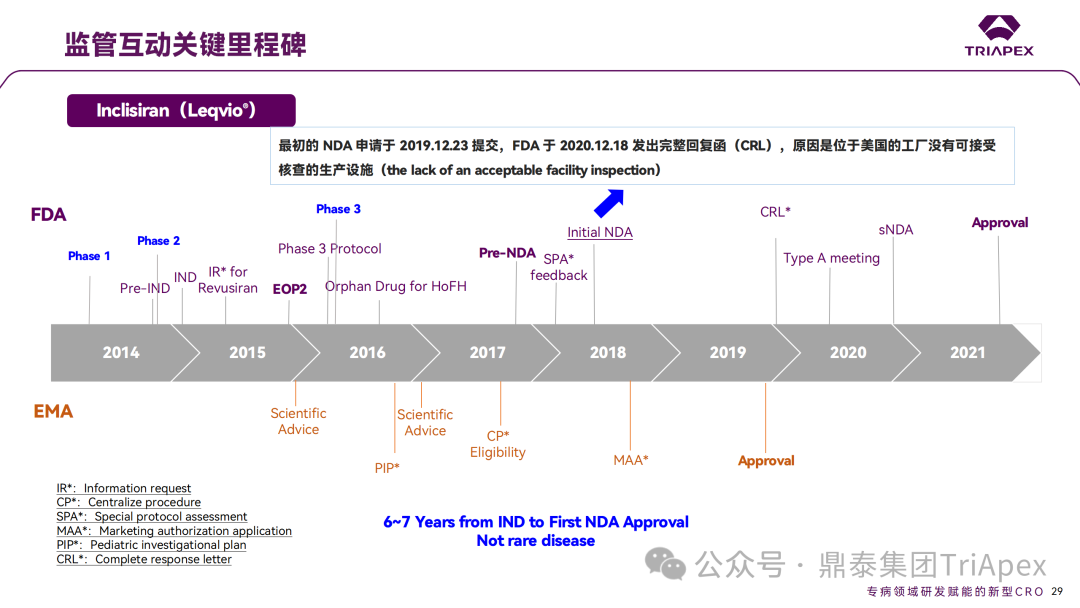


2、Pre-NDA 会议 在 Pre-NDA 会议中, FDA 就 Inclisiran 的临床药理学、临床安全性和有效性、优先审评等问题给出回复。 Pre-NDA 会议部分建议 在 Pre-NDA 会议召开后,申请人于 2019 年 12 月 23 日首次提交了 NDA,FDA 于 2020 年 12 月18 日发出 CRL 并表明不予批准,原因是位于美国的生产工厂没有可接受核查的生产设施。第二次 NDA 于 2021 年 7 月提交,并于 2021 年 12 月获得 FDA 批准上市。 3、Approval Letter 除 EOP2 会议、Pre-NDA 会议外,在 Approval Letter 中,FDA 就 ADA 检测及特殊人群相关研究做出要求。 Approval Letter

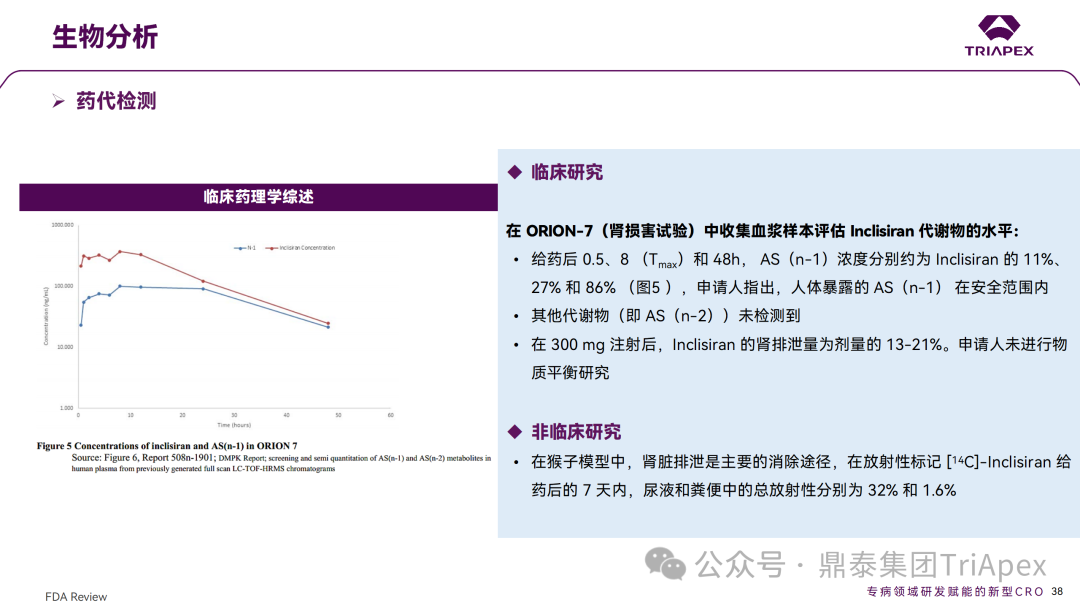
免疫原性检测与药代动力学 1、免疫原性和免疫毒性检测 在非临床研究中,采用 ELISA 法,分别在大鼠和猴的重复给药研究中评估了免疫原性。研究结果表明 ADA 的发生率较低,滴度较低,且 ADA 的发生与剂量无关,对 PD 结果无影响。 ≥100mg/kg 剂量下,在食蟹猴中观察到对 KLH 抗原延迟或缺乏(delayed or absent)IgM 免疫反应;300mg/kg 剂量下,观察到对 KLH IgG 免疫应答水平的降低,并且上述变化是可逆的。此外,Inclisiran 不会刺激小鼠或猴体内的促炎细胞因子,也不会激活猴体内的补体。 在 EOP2 会议(2017.04.06)中,申请人指出目前的临床研究中暂未在受试者中检测到 ADA。但在随后的一项 Ⅱ 期(ORION-3,2017.04.17开始)试验中,对 355 名受试者进行检测,结果发现治疗诱发的 ADA(Treatment Induced ADA)、持续性(Persistent)ADA、一过性(Transient)ADA 的受试者分别占 6.2 %、1.1%、5.1%,但治疗性 ADAs 患者均未发生相关的过敏事件。 在 3 项 Ⅲ 期(ORION-9、ORION-10 和 ORION-11)临床试验中,对 1830 名受试者进行了 ADA 检测,研究结果发现治疗诱导的 ADA 反应发生率为 4.9%,且滴度普遍较低,一过性 ADA、持续性 ADA 的发生率分别为 2.2%、1.7%。此外,申请人在 pre-NDA会 议中指出,ADA 的存在与 AE、LDL-C 和 PSCK9 水平的变化之间没有关联。 免疫原性检测结果 2、药代动力学 Inclisiran 代谢的酶是核酸外切酶。虽然在 GalNAc 丢失后 SS 链的 3' 端有代谢物,但由于潜在的药理活性,主要的代谢物是来自反义(AS)链的 5' 和 3'端 N-1 和 N-2 核苷酸的丢失,即 AS(N-1)3',AS(N-1)5',AS(N-2)3',AS(N-2)5'。 在非临床试验(大鼠和猴)中,AS(N-1)是主要代谢物,浓度比 Inclisiran低数倍。在猴子模型中,肾脏排泄是主要的消除途径,在放射性标记 [14C]-Inclisiran 给药后的 7 天内,尿液和粪便中的总放射性分别为 32% 和 1.6%。 在 Ⅲ 期临床 ORION-7(肾损害试验)中检测了血浆样本中 Inclisiran 代谢物的水平。研究结果发现,在给药后 0.5h、8h(Tmax)和 48h,AS(n-1)浓度分别约为 Inclisiran 的11%、27% 和 86%。未检测到其他代谢物,即AS(N-2)。在 300mg 注射后,Inclisiran 的肾排泄量为剂量的 13-21%。此外,申请人未进行物质平衡研究。 药代动力学检测结果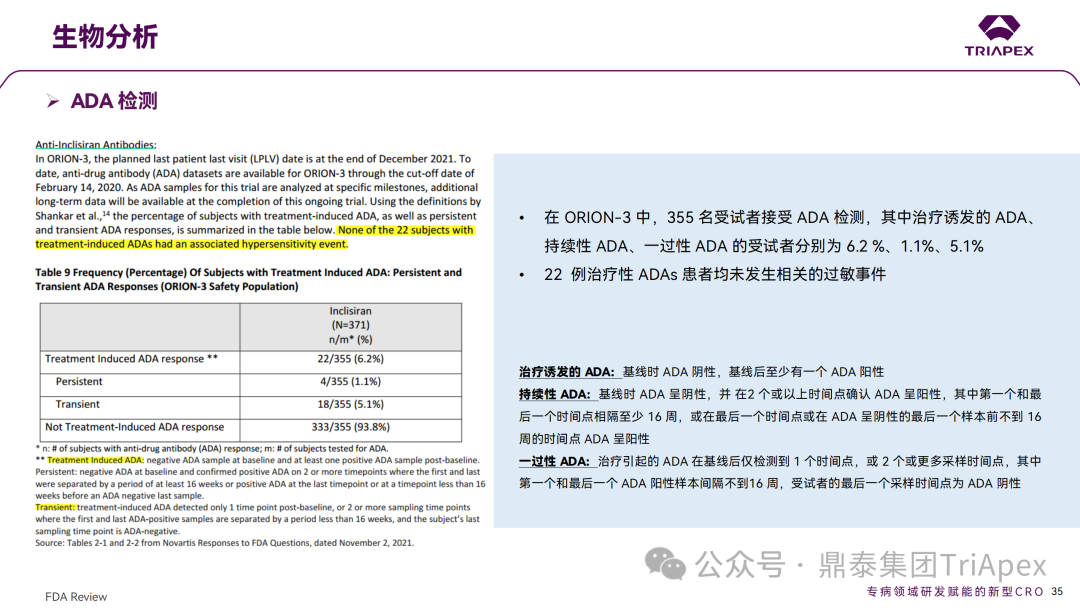


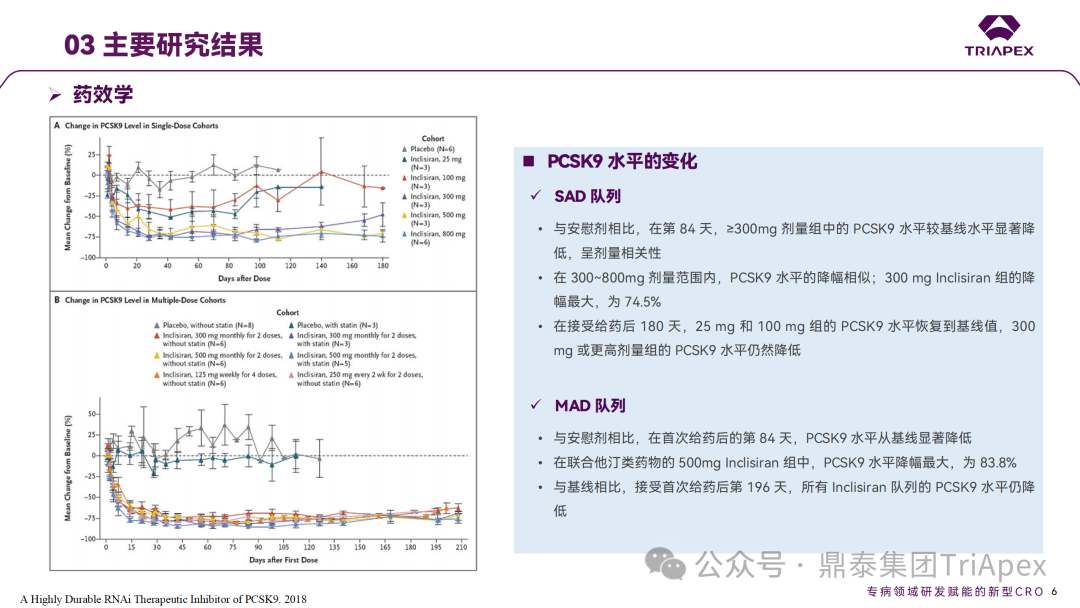
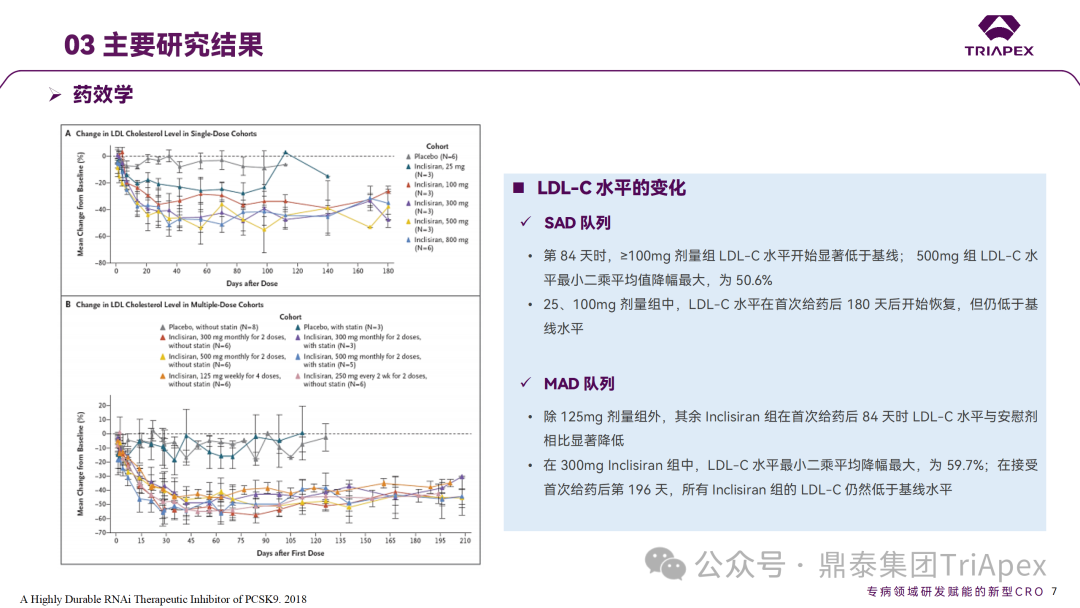
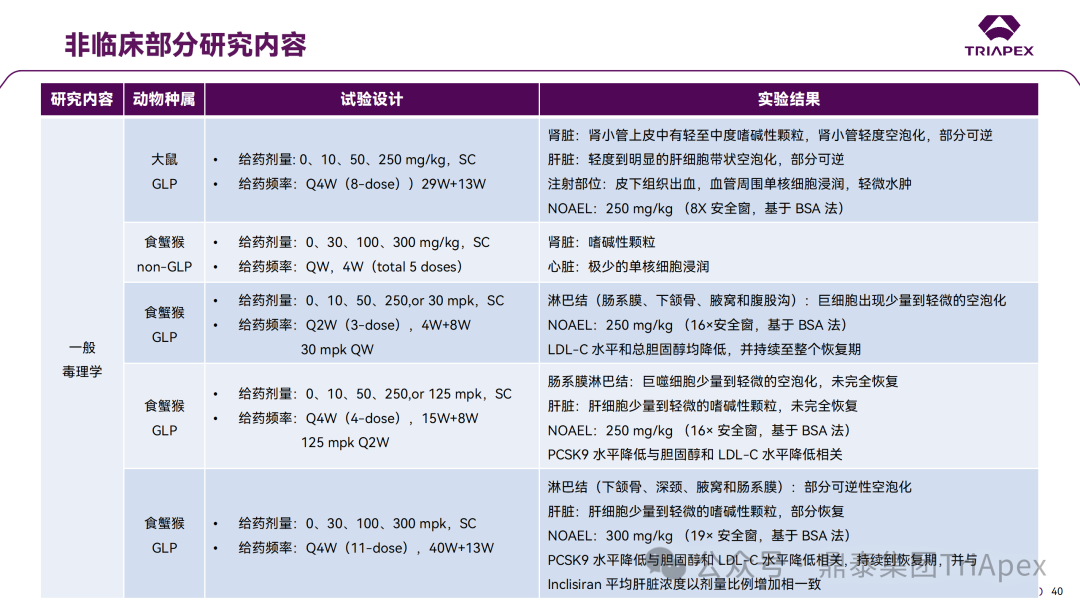
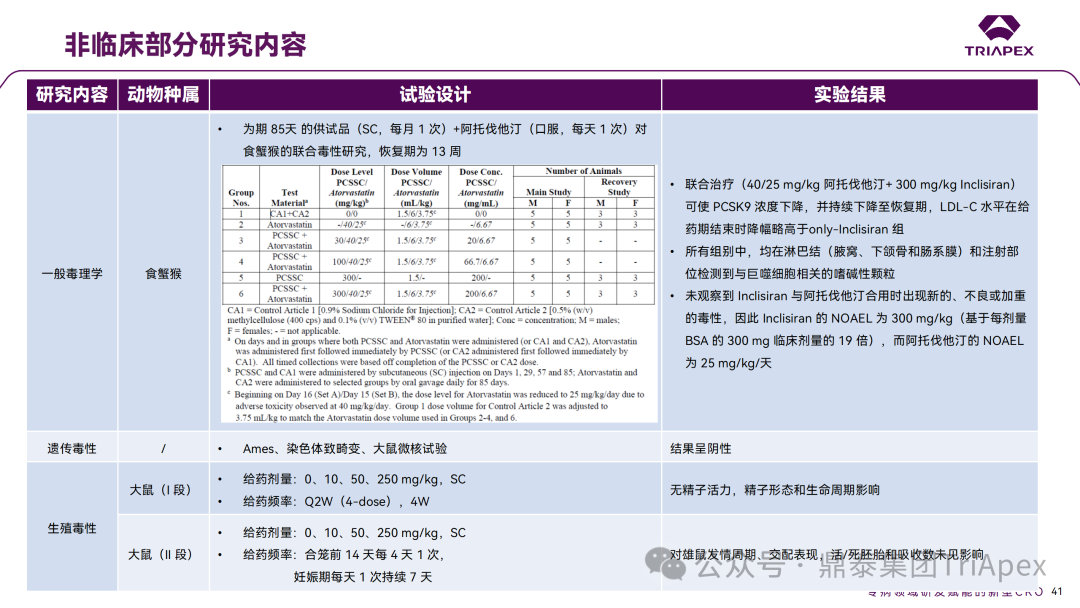
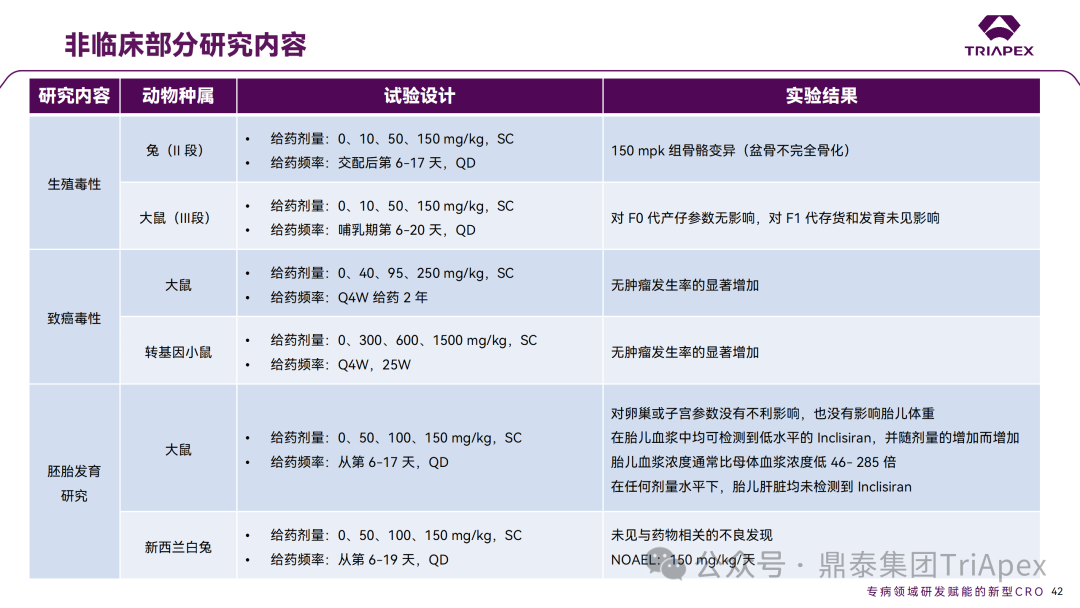
非临床与临床试验相关性分析 非临床研究与临床研究有着十分密切的关系。一方面,非临床研究方案的设计依赖于拟定的临床研究方案中的基本信息,包括 PK、PD 和安全性监测相关指标的确立;另一方面,非临床研究结果对临床给药方案的制定、安全性和有效性的评估等有重要指导意义。特别地,为支持本品与已上市他汀类药物的联合用药,开展了联合用药毒理试验。 非临床毒理研究内容 1、为临床药效学标志物提供参考 食蟹猴体内研究表明,单次给药后 Inclisiran 可剂量依赖性持续降低血清中 PCSK9 水平和 LDL-C 水平。在小鼠体内试验中观察到单次给药后 10 天 PCSK9 水平仍降低。在非临床研究中观察到的生物标志物(LDL-C、PCSK9)的变化,为临床研究中药物剂量设计、给药方案及药效标志物的选择提供了依据,因此在临床试验上,将 LDL-C 和 PCSK9 作为主要药效学指标。同时,非临床的研究结果,提示了 Inclisiran 在动物体内具有长期有效性,为临床研究提供了初步参考。 非临床 PD marker 与临床 PD marker 的相关性 2、为临床起始剂量的设计提供依据 非临床研究结果为 FIH 剂量设计提供了重要依据:在大鼠和猴重复给药 28 天毒性试验中,NOAEL 分别为 250mg/kg 和 300mg/kg(基于 BW 和 BSA 法),以此确定Ⅰ期研究 SAD 组受试者的起始剂量为 25 mg。 3、提示临床潜在不良反应 在 Inclisiran 非临床中发现的注射部位反应预示着临床研究中也会有潜在不良反应,并得以验证。 非临床毒性与临床 AE 的相关性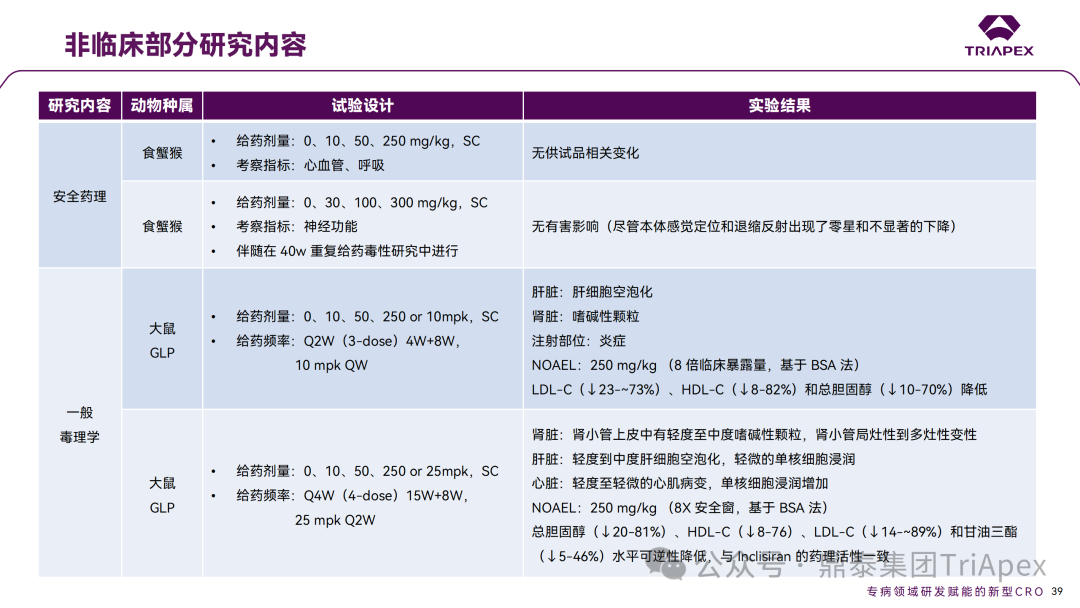


小结 作为唯一一款以降低胆固醇为目的的 siRNA 药物,Inclisiran 开启了血脂管理的新时代,实现了患者以更便捷、可依从、更安全的方式来进行血脂的长期管理,提升了患者的生活质量。当然,Inclisiran 的成功上市,离不开合理的非临床设计、科学的临床转化和高效的监管互动。借助合理的非临床设计,确定了临床起始剂量、临床生物标志物,为临床设计及结果解读提供强有力的数据支持;借助高效的监管互动,就产品开发中遇到的问题(如 ADA 的检测计划、PD 参数总体评估方法、整理临床试验设计等)与 FDA 积极沟通,并获得 FDA 的认同和建议。 Inclisiran 在慢性疾病领域的成功,吸引着越来越多的跟随创新和原创新靶点药物的开发,Inclisiran 的成功经验为 siRNA 药物的研发提供了宝贵的借鉴,这些策略和做法也为后续同类产品的研发提供了宝贵的经验和启示。
参考资料: [1]: Qureshi Z et al. Hypercholesterolemia: a literature review on management using tafolecimab: a novel member of PCSK9 monoclonal antibodies. Ann Med Surg (Lond). 2024 Mar 12;86(5):2818-2827. [2]: Henney NC et al. RNA Silencing in the Management of Dyslipidemias. Curr Atheroscler Rep. 2021 Sep 1;23(11):69. [3]: FDA审评报告 [4]: Gallego-Colon E et al. Statins and PCSK9 inhibitors: A new lipid-lowering therapy. Eur J Pharmacol. 2020 Jul 5;878:173114. Epub 2020 Apr 14. [5]: Rhee MY, et al. Ezetimibe and Rosuvastatin Combination Treatment Can Reduce the Dose of Rosuvastatin Without Compromising Its Lipid-lowering Efficacy. Clin Ther. 2019 Dec;41(12):2571-2592. Epub 2019 Nov 11. [6]: Chung, B.H.Y. et al. Rare versus common diseases: A false dichotomy in precision medicine. NPJ Genom. Med.2021, 6, 19. [7]: Lappin, J et al. Inclisiran. Pract Diab, 38: 41-43. 08 April 2021 [8]: EMA 说明书 [9]: FDA 说明书







 法规政策
法规政策 项目管理
项目管理


 Global
Global



 苏公网安备32011202001081
苏公网安备32011202001081