


药物致癌性试验是新药非临床安全性评价的重要研究内容。
自1995年以来,国际人用药品注册技术要求协调会(ICH)、经济合作与发展组织(OECD)、美国环境保护署(EPA)、美国食品药品监督管理局(FDA)、中国国家药品监督管理局(NMPA)等国内外机构陆续公布了一系列指导原则,用于规范和指导致癌性试验研究。当前,致癌性试验设计主要遵从 ICH S1A (1995)、ICH S1B(R1) (2022)及 ICH S1C(R2)(2008)的要求,分别从致癌性试验的必要性、试验设计及剂量选择三个方面进行阐述。
不同于其他非临床安全性研究,致癌性试验具有试验周期长、消耗大(每项试验需要约 500~700 只动物)、费用高,且试验设计(包括策略选择、具体试验剂量的设计等)复杂等特点,需要基于对已获得的信息(包括遗传毒性结果、拟用患者人群、临床拟用给药方案、动物和人体中的药效/药代数据、全身毒性研究数据等)的科学分析。建议在开展致癌性试验前,与监管机构进行及时沟通交流。
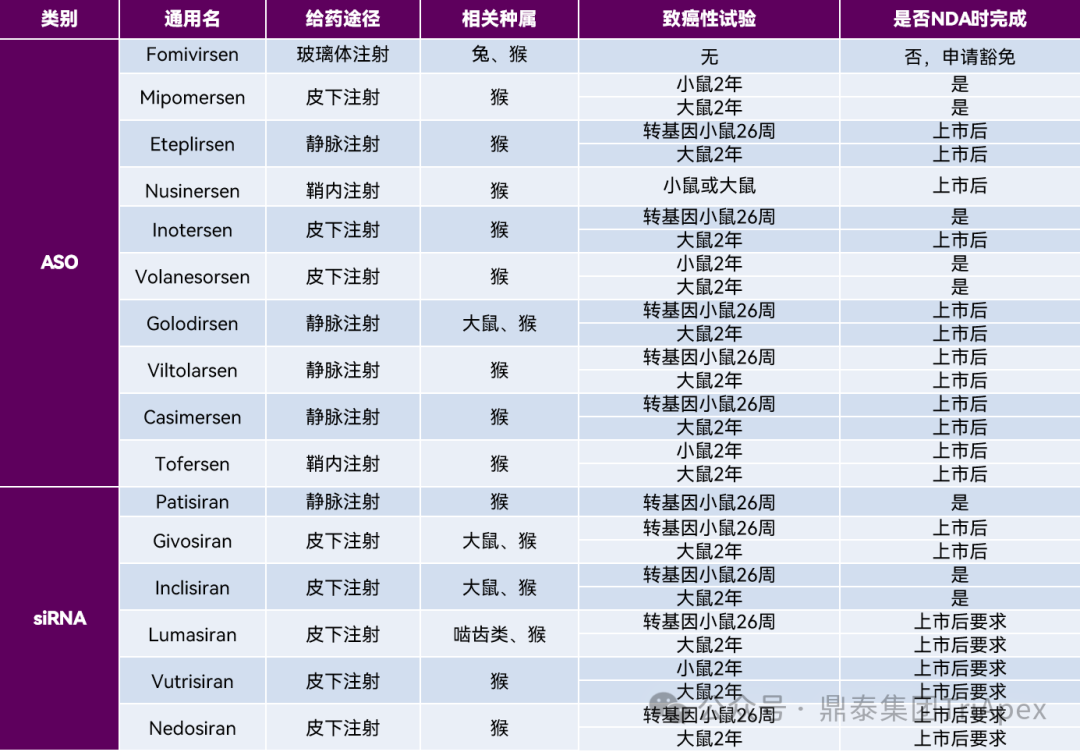
随着寡核苷酸药物研发热潮的兴起,很多拟用于慢性疾病的寡核苷酸药物符合 ICH S1A 中关于致癌性试验必要性的范围。鼎泰集团始终关注寡核苷酸类药物的研发进展和评价需求,除“他山之石”系列文章(点击阅读)、非临床药效学研究、杂质控制和相关指导原则解读外,也对于致癌性试验的开展情况进行了汇总和解析(详见下表)。
从公开资料来看,多数产品采用双种属(小鼠/转基因小鼠和大鼠)进行致癌性试验,或被要求在上市后开展致癌性试验。其中,有3款产品的致癌性试验设计比较有趣:
Patisiran,仅采用转基因小鼠进行为期 26 周致癌性试验,究其原因与大鼠重复给药 26 周毒性试验末次给药未见药物检出(免疫原性导致)相关;
Nusinersen 和 Fomivirsen,均为局部用药,申请人从给药途径的特殊性和技术难度、系统暴露的局限性、作用机制、遗传毒性试验阴性结果、重复给药未见癌前病变提示等角度申请豁免。不过,对于同一问题,监管视角的评估往往更为重要。审评员要求 Nusinersen 在上市后补充单一种属的致癌性试验;而 Fomivirsen 则顺利豁免。进一步调研发现,Nusinersen 在临床人群中具有明显的系统暴露,这可能是监管机构要求增加致癌性试验的深层次原因。
综上可见,致癌性试验的实施通常是在遵照指南的基础上,基于临床风险评估进行科学合理的设计,为产品的临床应用安全保驾护航。
以下是鼎泰团队总结的关于已获批寡核苷酸类药物致癌性试验开展情况。
鼎泰团队始终秉承为专病领域赋能的愿景,以专注科学评估、助力创新转化、提升生命质量为使命,顺应药物安全性评价的需求。现已组织开展致癌性试验并成功获得开展致癌性试验的 GLP 资质,试验设施、人员及试验体系均通过监管机构核查。在此期间,积累了丰富的背景数据,为开展致癌性试验打下了坚实的基础,可以满足工业界对致癌性试验开展的需求。
恰逢近期有业内资深同行对药物致癌性实验的一般考虑进行了汇总整理,在征得原创作者同意的前提下,鼎泰团队对下述内容进行转发,以飨同道,共享参考。
肿瘤是以细胞不可控的生长和传播为特点,并最终导致机体的死亡。肿瘤的发生既可以来自于外部因素如辐射、致癌化合物暴露、微生物感染或一些物理作用,也可以来自内部如基因突变、激素、免疫功能抑制等。药物潜在致癌机制也是多种多样,所以需要在上市前对致癌风险进行评估,并根据情况完成相关研究。
先看下监管机构对药物致癌性试验的要求。对于用药周期达到 3 个月或更久的药物,FDA 要求在上市前提交致癌性研究资料。日本和欧洲则要求 6 个月及以上的药物提供。后续 ICH 将这一药物暴露周期也定在 6 个月。当然,如果在短期暴露或非频繁给药情况下,出现了致癌方面的担忧,如遗传毒性结果阳性,构效关系提示致癌风险,短期重复给药试验发现癌前病变或同类产品已观测到人类致癌风险等,那就不能按照 6 个月这一条件决定致癌性试验开展与否了。
内源性多肽、蛋白或其类似物通常不需要开展致癌性试验。但是,如果药物与内源性成分生物学作用迥异,或药物经过修饰导致结构与内源性成分完全不同,或药物暴露量较内源性成分高很多,则还是需要进行致癌性评估。
并不是所有药物均需要开展 2 年大鼠致癌性研究。ICH S1 认为如果从药物靶点、信号通路、毒理学研究等综合分析,认为药物致癌风险很低,或者明确有致癌风险,则 2 年大鼠致癌性试验开展的意义并不大。证据权重分析(WoE)分析维度如下图所示,从靶点生物学作用、次要药效学、重复给药毒性研究结果、对激素的影响、遗传毒性和免疫调节作用,综合评估可能的致癌风险。对于“likely” 和 “unlikely”两种情况,额外开展 2 年大鼠致癌研究并无意义。对于不确定的情况,则需要开展大鼠 2 年致癌性试验。
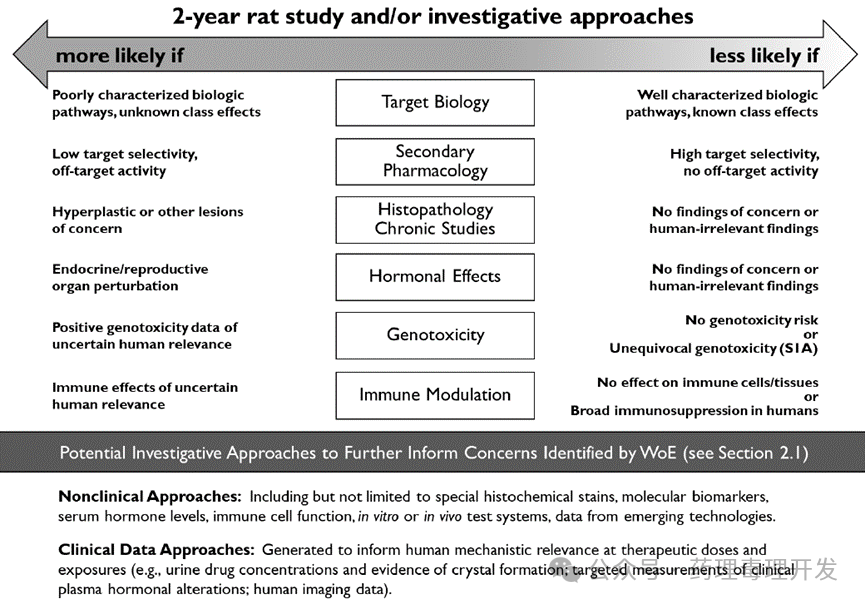
不过小鼠致癌性试验往往通常还是要开展的,即使 WoE 认为药物人体潜在致癌风险为“unlikely”。根据动物“3R”原则,建议优先选择转基因动物模型。当然,某些情况下,小鼠试验也可以不进行,比如 WoE 提示药物致癌风险低,且小鼠中达不到人体治疗剂量下的暴露水平。又如 WoE 提示药物很可能是人体致癌物,小鼠试验也不适合继续开展。
种属选择
致癌性试验常规使用的是两种啮齿类动物组合,小鼠和大鼠。小鼠最常用的种系是 CD-1,其他品系如 B6C3D1、CF-1、NMRI、C57B1、Balb/c 则用的相对较少。大鼠最常用的是 SD 大鼠,某些企业喜欢用 Hans Wistar、Fischer344(F344),Long Evans 和 CFE 用的很少。那如果大鼠和小鼠都不合适呢,仓鼠(hamster)作为替补队员就上场了。
都是啮齿类种属,为啥要用两种动物进行致癌评估呢?因为基于过往认知,没有任一种属能够充分预测人体致癌风险。如果两种种属均未发现致癌风险,结果的可靠性要强于单一种属。关于两个种属之间致癌结果是否一致问题,不同数据来源出入挺大的,Purchase、Haseman、Gold 团队公布的这一数据分别是 82%、73%、30-40%。无论如何,任一啮齿类种属致癌性结果并不能用于预测另外一种啮齿类动物,所以双种属致癌性试验组合还是会继续延续。
种属选择的考量因素有很多,除了传统的相关种属判定,动物自发肿瘤、生存期、充分的背景数据等也需要作为选择标准。而且,不同啮齿类品系各种类型的肿瘤发生率区别很大。
以最常用的 SD 大鼠为例,老年雌性 SD 大鼠有很高的乳腺癌发病率,且很多动物因为这个原因在致癌性试验中的第二年死亡;
F344 大鼠虽然乳腺癌发病率低很多,但睾丸肿瘤的发病率接近 100%。
自发性肿瘤会很大程度上影响致癌性结果的判定,首先如果受试物引发的某种肿瘤在该种属中自发率很高,则会有很高的背景噪音,评价难度增加。其次,即使结果显示较背景数据高很多,也可能是这个种属对该肿瘤比较敏感,毕竟有很高的自发率,对于能否预测人体情况,也很难判定。
除自发性肿瘤,动物生存期也需重点考虑,正常需要考察药物在其正常生命周期暴露后引发的潜在致癌风险。如果动物在试验期间死亡,则不能提供足够的肿瘤形成周期,整个试验的灵敏度也降低,假阴性的概率则会增加。另外,还需要积累充分的背景数据,辅助致癌结果的判定,比如 CD-1 小鼠脾脏、肝脏、肾脏、甲状腺、肾上腺有比较高的淀粉样变性自发病变。
动物饲养
致癌性试验以周期长和花费高著称。这期间动物护理非常重要,也会极大影响试验质量。各种物理、生物学因素引入的风险需要仔细评估。
物理因素包括光、温度、相对湿度、通风、噪音、饮食、居住条件等,生物因素诸如细菌和病毒可能引起感染和其他疾病。例如:
光照持续时间、强度和光源质量会影响动物肿瘤发病率;
光强过大会引起动物眼部损伤;
居住在高排和侧面笼位的动物受影响最大;
室温异常会使小鼠皮肤肿瘤发生率增加;
相对湿度改变则会影响动物饮水和摄食;
低的湿度还会引发动物“ringtail”(环尾)。
啮齿类动物致癌性试验中的理想饮食要营养充分,但又不能营养过剩。FDA 研究发现,现在大鼠的寿命在降低,与饮食有很大关系。
饲养笼的类型和垫料也会影响小鼠皮肤肿瘤的发病率和发病周期。DePass 团队 1986 年研究显示,不锈钢笼子和聚碳酸酯笼盒饲养的小鼠,后者肿瘤发生的更早,且发生率也更高。
给药组数量
对照组数量
与其它毒理试验通常仅设置 1 组对照不同,致癌性试验常用的是设置 2 个对照组,每组动物数相同。当然,也有设置 1 个对照组,动物数量与给药组一样的设计。还有一种比较少用的设计,设置 1 个对照组,数量是给药组的 2 倍。
使用两组对照的优势是可以评估两组之间的自发肿瘤的变异情况。假如两组间无差异,数据可以合并使用,这种情况下两个对照组和一个对照组的设计就没有区别。假如两组间有差异,则必须将给药组的数据与两个对照组分别进行对比。那如果对比完发现,给药组某个肿瘤发生率比其中一个对照组高,跟另外一个对照组相似怎么办呢?这就需要结合历史背景对照数据进行分析。
剂量设计标准
剂量设计本来就是毒理研究的重要考量点,对于致癌性试验那就更是重中之重了。毕竟,急毒或短期剂量探索试验剂量设计不合理还有后续重复给药毒理试验补救,致癌性试验就没有这种机会了。尤其当致癌性试验后期才暴露这一问题,就更讨厌了。
大部分药企剂量设计的原则是,高剂量为最大耐受剂量(MTD),低剂量是最大临床推荐剂量(MRHD)的 1-5 倍,中剂量为两个剂量之间的几何平均值。ICH(1995)对 MTD 描述“that dose which is predicted to produce a minimum toxic effect over the course of the carcinogenicity study, usually predicted from the results of a 90-day study”。MTD 如果设计合理,首先不会影响动物的生存期,其次对动物体重增长有中等影响,并产生一定的毒性症状。对于没有毒性的药物,按照传统 MTD 的高剂量设计思路,高剂量和低剂量可能会差好几个数量级。有些指南不建议高低剂量相差超过 10 倍,这种情况下可以将高剂量定为 MRHD 的 100 倍。其实,按照低剂量是 MRHD 的 1-5 倍计,已经有些超出 10 倍的要求了。对于掺食给药的情况,日本监管机构规定,即使 MTD 还没达到,高剂量组药物在饮食中的比例不应超过 5%。
当然,剂量选择时还要考虑药物药代动力学特点,不建议选择的高剂量在排泄方式上与低剂量有很大不同。代谢角度的考量也是类似的,不建议高剂量产生的代谢产物与低剂量有很大不同。另外,如果药物在某个剂量已经达到最大暴露,也不建议继续提高给药剂量。
每组动物数
建议每组每性别至少 50 只。大部分企业选择这一最低数量,也有企业选择 80 只/性别/组。决定动物数量多少的核心要素是在试验终点时,保证有足够数量的动物用于致癌性评估。尤其是每天灌胃给药的情况,有可能因对食管造成的损伤或药物误入气管,引起动物非正常死亡,更需要每组多设置一些动物。还有些情况是致癌性试验和慢性毒性试验合并开展,需要在第 6 和 12 个月时处死一些动物,用于潜在的一般毒性评估,也需要每组的样本量更大。
给药途径
最常见的两种给药途径是掺食和灌胃。其它给药途径包括皮肤用药、掺入饮水、注射给药,视临床拟用途径而定。如果候选药物临床给药途径不只一种,通常建议掺食给药。掺食的好处是节约人力,毕竟每天灌胃几百上千只动物是不小的工作量。另外,有研究显示掺食不会像灌胃那样,过高估计 MTD。不过,如果药物在啮齿类饮食中不稳定或影响动物正常摄食,就另当别论了。掺食另外一个缺点是给药剂量只能靠动物体重和摄食数据计算,不像灌胃给药剂量那么准确。
试验周期
通常,大小鼠致癌性试验周期都是 2 年。偶尔也有大鼠周期延长至 30 个月,小鼠周期缩短至 18 个月。出现这种区别的原因是过去认为大鼠的寿命比小鼠更长。不过,很多研究已经证实 SD 大鼠和 CD-1 小鼠的生存期都是 2 年左右。其它品系,如 F344 大鼠和 B6C3F1 小鼠,也是一样的 2 年寿命。
生存期
为确保能观察到某些迟发肿瘤(比如某些弱致癌物),受试药物需要尽可能久的暴露在动物的生命周期中,这就需要动物要活的足够久。如果动物在早期出现非肿瘤原因导致的死亡,会影响试验的质量和试验目的的达成。很多时候是致癌物的致癌潜力越弱,观察到肿瘤发生的时间越晚。致癌物的剂量越低,观察到肿瘤发生的时间同样也晚。所以,动物生存期是致癌性试验需要重点关注的。关于动物最低生存期,没有统一要求。FDA Redbook II Draft Guideline 建议大鼠、小鼠或仓鼠要给药 24 个月。EEC guideline 则认为对照组存活率达到 20% 时,就可以结束试验。日本监管机构则认为对照组或低剂量组存活率还剩 25% 时,就可以结束研究。
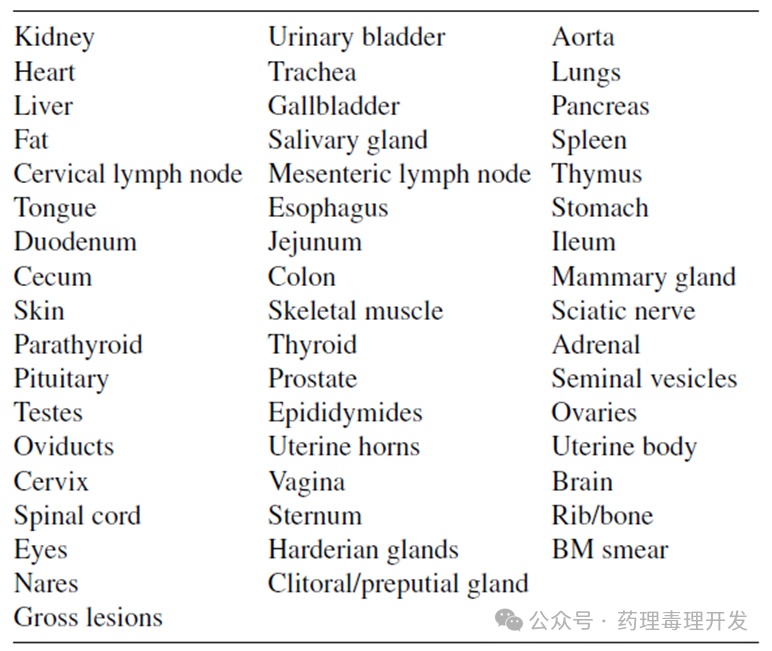
试验终点检查
致癌性试验的终点比慢性毒理试验更为聚焦,终点指标更少。关键终点包括病理学、体重、生存期、临床病理、摄食量。标准病理组织清单如下:
致癌数据的评估需要关注以下变化:1)剂量-反应关系;2)肿瘤常见器官中的间变性肿瘤变化趋势;3)肿瘤出现时间更早的趋势;4)存在癌前病变的迹象。
致癌性试验阳性结果的判定通常有3个标准,分别是:1)常见肿瘤出现统计学意义的增加;2)肿瘤出现的时间明显提前;3)虽然无统计学意义,但出现非常罕见肿瘤,也判为阳性。前两个是基于统计学分析进行判定。
4 款常见的转基因小鼠
致癌性试验的常规组合是大鼠和小鼠 2 年给药致癌研究。但这种动物全生命周期给药的方式也常常遭受质疑。因此,1997 年 ICH 提供了一种中期或短期小鼠试验,以替代小鼠 2 年全生命周期的研究。之后,FDA 对于基于转基因小鼠开展中等周期致癌性试验也逐渐接受。当然,转基因小鼠有充分的验证数据。目前验证最为充分,使用较多的是 4 种转基因小鼠模型,分别是 TSPp53+/-、Tg.AC、Hras2 和 XPA-/-。每种模型都有其独特的特点。
1
Tg.AC 小鼠模型
这是最早被开发出来的转基因致癌小鼠模型。早在 1990 年就被报道用于小鼠皮肤致癌性试验。这个小鼠体内 11 号染色体上有 4 个拷贝的 v-H-ras 致癌基因,并与启动子 ζ-globin 基因进行了融合。改造的基因与小鼠皮肤的快速成瘤有关。将一些潜在致癌物用于皮肤表面可以较短时间诱导出乳头状瘤。40% 的乳头状瘤会进一步发展为皮肤恶性肿瘤,主要是鳞状细胞癌和肉瘤。
该模型在美国被美国环境健康科学研究所(NIEHS)和美国国家毒理学计划(NTP)首次用于替代传统致癌性试验。这一试验中有 40 种化合物被用于致癌性测试,并最终发表,算是开了短期试验替代传统长期致癌研究的先河。无论皮肤受试物还是需系统给药的化合物,大多数使用 Tg.AC 小鼠模型开展的致癌性试验均采用皮肤给药。这就要求对皮肤的处理要比较精细、小心,因为皮肤损伤也会形成乳头状瘤。
这个模型的问题是,并不是所有拟系统给药的药物均适合经皮途径给药。这也是为什么国际生命科学学会(ILSI)启动了一项在该模型中经皮和系统双给药途径研究计划。另外,该模型还会发生对阳性对照致癌物不响应的情况,原因与 ζ-globin 启动子重排有关。不过,据称这一问题已得到解决。
2
Tg.rasH2 模型
该模型由日本实验动物中央研究所(CIEA)研发,并于 1990 年首次对外披露信息。该模型小鼠的改造策略是将 5 或 6 个拷贝的人 H-ras 原癌基因以串联的形式插入基因组。Tg.rasH2 小鼠非常稳定,自模型开发成功以来,从未出现过响应缺失的情况。该模型编码的开关蛋白与 Tg.AC 小鼠类似,但不同的 Tg.rasH2 小鼠转入的基因在所有器官和组织均表达,不仅限于皮肤。
采用 N-甲基-N-亚硝脲(MNU)给予该小鼠,可快速诱导前胃乳头状瘤的出现。故,MNU 后续常作为 Tg.rasH2 小鼠致癌性研究的阳性对照物。通常,采用 Tg.rasH2 小鼠开展的致癌性试验,6 个月给药周期就足够了。因为这个节点动物已经出现各种自发性肿瘤,如脾脏血管肉瘤、前胃和皮肤乳头状瘤、肺腺癌和 Harderian gland 腺癌以及淋巴瘤。与其它致癌小鼠模型相比,这个模型特点是对致癌物的反应非常强烈,直接表现是一旦接触致癌物,阳性反应出现的更早、发生率也更高。具体机制尚不清楚。
虽然 Tg.rasH2 对少部分化合物给出的结果模棱两可,但不可否认的是,大多数非致癌物均能在该模型得到客观、正确的鉴定。Tg.rasH2 是目前转基因致癌小鼠模型中最受欢迎的。
3
P53+/- 小鼠模型
TSP P53+/-杂合子敲除小鼠模型由美国开发。P53 基因是最重要的肿瘤抑制因子之一,恶性肿瘤中经常能看到该基因的突变。当 DNA 链受损时,该因子会激活多个基因,这反过来会导致 DNA 修复时细胞周期的停滞,或者导致细胞凋亡(程序性细胞死亡),从而清除受损细胞。之所以使用杂合子,是因为纯合子在出生后几个月内自发性肿瘤的发生率非常高。杂合子在长达 12 个月内的肿瘤背景发病率较低,但在这段时间内,如果动物给予致癌物,进一步发生 p53 基因突变事件的可能性很高。从而导致抑癌功能的完全丧失,恶性肿瘤发生风险升高。
最早是由美国 NIEHS 采用该模型进行了致癌性研究,希望作为传统致癌体系的备选之一。结果发现该模型对致突变致癌物苯和甲基苯胺是敏感的,6 个月内即出现阳性结果。但是,ILSI 后续研究发现,该模型 6 个月的给药周期可能是不充分的。尤其是苯,6 个月出现阴性或模棱两可的结果,9 个月才出现比较确定的阳性结果。另外,非致突变致癌物和致突变非致癌物在该模型中均呈现阴性结果。
P53+/-小鼠是美国最为流行的转基因致癌模型。
4
XPA-/-小鼠模型
XPA 基因的两个等位基因均通过 ES 细胞中的同源重组而失活,导致跨外显子 3 和 4 的基因纯合缺失。该基因编码的蛋白可以通过 NER 通路,检测并修复 DNA 损伤。XPA-/-小鼠模型仅残留了 2%-5% 的 NER 活性。荷兰国家公共卫生及环境研究院(RIVM)1995 年首次发表了该模型的数据。发现,XPA-/-小鼠暴露于 IV-B 放射或 7,12-二甲基苯[α]蒽后可快速诱导皮肤肿瘤的发生。经口给予其它致突变致癌物后,可以诱导多种体内肿瘤的发生。早期研究认为 6 个月足以观察到肿瘤发生,后续更多试验表明 9 个月对某些阳性药物才更为充分如 B[α]P, 2-AAF. XPA-/-小鼠对 UV、基因毒性致癌物和非基因毒性致癌物都是敏感的。而且,非致癌物在该模型中未见假阳性结果的出现。
还有一种 XPA-/-P53+/- 双转基因小鼠,这个动物对致癌物更为敏感。比如 DES(己烯雌酚)开展的一项研究中,XPA-/-P53+/- 双转基因小鼠出现 7 个动物转移性骨肉瘤,XPA-/-小鼠仅 1 只。但这一模型有可能过于敏感,出现致癌阳性结果与人体风险之间的相关性存疑。
ICH S1B(1997) 已经将以上模型写入了指导原则中,用于替代传统长期小鼠致癌性试验。当然,传统大鼠致癌性试验还是要开展的。FDA 对转基因模型也是持开放态度。甚至, FDA CBER 对于遗传毒性结果阴性,且从作用机制角度没有太多担忧,单一种属转基因动物模型,也有接受的案例。







 法规政策
法规政策 项目管理
项目管理


 Global
Global



 苏公网安备32011202001081
苏公网安备32011202001081