


往期内容《EMA 寡核苷酸药物指导原则,还远吗?》(点击阅读全文)分享了欧洲药品管理局 (European Medicines Agency , EMA) 为推动 RNA 药物研发和指南制定而进行的一系列举措。如文章中所预期的,EMA 于近日(2024 年 7 月 14 日)发布了 基于 2022 年 6 月 EMA 发布的


点击查看大图
全文共:7858 字 11 图 预计阅读时长:20 分钟
1.适用范围 该指南目的是规定为获得上市许可而进行的合成寡核苷酸(现有或新化学实体)在开发、生产和控制方面所需的信息类型,其中还包括一章临床试验申请: 特定方面的要求:合成寡核苷酸被完全或部分排除在 ICH Q3A/B (VICH 72 GL10/GL11)、ICH Q6A/B (VICH GL39/GL40) 和 ICH M7 (EMA/CVMP/SWP/377245/2016) 的范围内。针对合成寡核苷酸的生产过程、表征、规格和分析控制,指南提供了具体指导,这些内容未被 兽用产品的适用性:尽管兽用产品不在 ICH 指南的范围内,但考虑到灵活性和允许申请人采用不同的产品开发方法,本指南中概述的原则也可能适用于兽用产品。 排除范围:mRNA 实体不在本指南的范围内。 2. 法律依据和相关指南 本节提到了多个法规和指南,覆盖了稳定性测试、分析方法验证、杂质控制、溶剂残留检测、元素杂质管理、质量标准制定、制药开发和质量风险管理等多个方面,为合成寡核苷酸的生产提供了全面的法律和技术框架。 指南建议,应与第 2001/83/EC 指令附件一的介绍和一般原则以及第 2019/6 号条例 (EU) 附件二的修改版人用药品和兽用药品,以及所有其他相关的欧盟和 (V)ICH 指南一起阅读。
3. 原料药 ▶ 基本信息(3.2.S.1) 需要详细记录合成寡核苷酸的药品名称(3.2.S.1.1)、结构(3.2.S.1.2)和基本性质(3.2.S.1.3),包括但不限于分子式、分子量、立体化学讨论等,以确保对其化学特性的全面了解。 ▶ 生产(3.2.S.2) 详细描述了合成寡核苷酸的生产过程,包括生产商信息(3.2.S.2.1)、生产工艺和工艺控制(3.2.S.2.2)、物料控制(3.2.S.2.3)、关键步骤和中间体控制(3.2.S.2.4)、工艺验证和/或评估(3.2.S.2.5)以及生产工艺的开发(3.2.S.2.6),确保了生产过程的可追溯性和可控性。 ▶ 特性鉴定(3.2.S.3) ● 结构和理化性质(3.2.S.3.1) 通过多种技术手段,如质谱分析、核磁共振(NMR)、紫外光谱(UV)、圆二色谱(CD) 和红外光谱(IR) 等,对合成寡核苷酸的结构和特性进行全面表征,包括一级、二级或三级结构的确认,以确保其结构完整性和生物活性。 元素分析:可以通过元素分析(例如燃烧分析或电感耦合等离子体发射光谱法 [ICP-OES] 测定磷和钠含量)进一步确认结构。 NMR:推荐将 NMR 作为特性鉴定的一部分,以阐明核碱基、糖和骨架组成、成分和核苷酸的连接性。 核苷立体化学:需要讨论核苷立体化学,包括源自其核苷前体的亚磷酰胺起始物料的立体化学纯度,以及在寡核苷酸合成过程中潜在的异构化倾向。需要讨论立体化学对生物/药理活性的影响。 生物测定:对于通过与互补链退火发挥作用的反义寡核苷酸或 siRNA,通常不需要生物测定来进行特性研究或常规放行。对于通过选择性结合靶分子而发挥作用的适配体,因其生物活性严重依赖于三级结构,期望对其属性(例如,对生物靶标的结合)进行特性研究。 List of characterisation techniques used for oligonucleotides (example table) 物理化学特性:合成寡核苷酸的物理化学特性的表征可能包括溶解度和吸湿性研究、等电点(pI) 的测定以及热重分析 [如差示扫描量热法(DSC) 和热重分析(TGA)]。形态学可以通过粉末X射线衍射(PXRD) 和/或 DSC 进行检查。 ● 杂质研究(3.2.S.3.2) 纯度是寡核苷酸的重要关键质量属性(CQA)。杂质通常分为两类:产品相关杂质和非寡核苷酸杂质。与产品相关的杂质包含寡核苷酸序列的结构元素;非寡核苷酸杂质包括工艺试剂及其可能的副产物、残留溶剂、元素杂质和可能的致突变杂质。 产品相关的杂质不同来源 起始物料:起始物料中的反应性杂质可能在寡核苷酸合成过程中引入序列。 生产过程中形成:生产过程中由于不完全或非预期反应产生。 生产或储存过程中的降解:生产或储存过程中产生的降解产物。 某些与产品相关的杂质可能是这些来源的综合影响引起的结果。 起始物料相关的物质 起始物料中的反应性杂质,通常会在最终的原料药中持续存在,其水平取决于起始物料中杂质的含量及其在寡核苷酸链中的使用频率。下游处理和纯化步骤通常无法去除这些杂质。严格的质量标准应设定用于起始物料中的反应性和关键杂质。 生产过程中形成的物质 磷酸二酯(PO) 杂质、立体异构体、缺失序列和截短序列、插入序列、含有碱基修饰的寡核苷酸、交叉连接寡核苷酸、双链寡核苷酸中的单链残留物、双链寡核苷的残留单链等。 产品相关杂质的界定要求(Qualification requirements) Class I:与主要代谢物结构相同的杂质,无需进一步鉴定。 Class II:仅含天然存在的核酸结构元素的杂质,无需进一步鉴定。 Class III:序列变异的杂质(如 n-1 或 n+1 杂质),需单独识别并鉴定超过 1.5% 阈值的杂质。 Class IV:含有非亲本寡核苷酸或非天然核酸结构元素的杂质,若超过 1.5% 需进行鉴定。 ▶ 原料药的质量控制(3.2.S.4) ● 质量标准(3.2.S.4.1) 合成寡核苷酸不在 ICH Q6A 的范围内,但质量标准的制定可以考虑 ICH Q6A 中概述的原则(兽用产品参考 VICH GL39)。指南中列出了寡核苷酸质量标准中的典型质量属性。对于双链寡核苷酸,应同时使用非变性方法(以测量单链残留物)和变性方法测试纯度。 对于适配体,应在原料药或制剂的质量标准中包括生物活性测试。 ● 分析方法(3.2.S.4.2) 阐述了分析方法开发以及开发过程中分析方法变更相关的内容。包括鉴定、纯度、分析、开发过程中分析方法的变化。 其中,指南指出,应明确定义测定方法及其计算方式。可以通过 UV 或通过基于重量的测定法与使用 HPLC 的已知纯度和浓度标准品进行测定,并通过 UV 吸收检测。结果计算还可以使用消光系数。在寡核苷酸的开发阶段,可能会对分析方法进行不同程度的变更,不论是重大或微小变更,应对开发期间进行的变更进行讨论。 ● 分析方法验证(3.2.S.4.3) 在药物活性成分质量控制中所采用的分析方法必须经过全面验证。通常,原料药分析测试的验证应遵循现行指南(如 ICH Q2、VICH GL1 和 GL2)的要求。具体而言,需要提供色谱图以及相关的 MS 数据,以证明寡核苷酸相关杂质与原料药峰之间的分离程度足以允许进行峰积分,从而确保分析结果的准确性和可靠性。 ● 批次分析(3.2.S.4.4) 应汇总用于非临床和临床研究以及支持原料药质量的寡核苷酸批次的分析数据。推荐以表格形式呈现这些信息,以便于清晰理解。对于每一批次,除了分析测定外,还应提供生产日期、批号、批大小(以质量或摩尔浓度表示)、规模(实验室/试点/商业)、合成路线(是否商业化)、生产地点以及批次用途等详细信息。 从早期阶段到最终的商业化批次的开发过程中,质量标准往往会发生变化。应解释并合理化在早期开发和试验批次/商业批次中获得的结果差异。在开发过程中改进分析方法可能导致在试验批次/商业批次中观察到新的杂质或杂质的组合。在这种情况下,应进行批次分析数据比较,并讨论这些新杂质(组)的鉴定必要性。 ● 质量标准制定依据(3.2.S.4.5) 预期的质量标准应基于非临床、临床研究以及支持原料药的批次数据,结合对生产过程的充分理解及可能影响寡核苷酸关键质量属性的因素。对于鉴定测试,至少需要基于两种互补技术(见 3.2.S.4.2)的规格,其中一种技术为序列确认测试。对于残留溶剂、元素杂质、细菌内毒素和微生物学等参数的限制,应依据适用的 EU/(V)ICH Guidelines, Ph. Eur. Or European legislation 进行合理化的论证。寡核苷酸相关杂质的要求,包括鉴定方法,应按照 3.2.S.3.2 和 3.2.S.4.2 中指示的方式进行合理说明。这确保了质量标准的设定既科学又合理,能够有效控制产品质量。 ▶ 对照品或参考品(3.2.S.5) 讨论了对照品或参考品的使用,为活性物质的质量控制提供了基准。寡核苷酸通常是非常具有吸湿性的粉末,因此在存储和分析过程中,应当参考相关标准,采取适当的预防措施防止吸湿。如果适用,参考标准的来源应简要说明(例如,根据商业过程合成的批次)。 对于双链寡核苷酸,需要正义链、反义链和原料药本身的自制品或参考品。 如果为了单链寡核苷酸的鉴定测试(例如熔解温度)而使用互补链,应进行描述。 ▶ 包装系统(3.2.S.6) 提供了关于包装系统的信息,以保护活性物质免受外界环境影响。 ▶ 稳定性(3.2.S.7) 稳定性研究包括稳定性总结、结论、批准后稳定性研究和承诺,以及稳定性数据,以评估活性物质在不同条件下的稳定性。 ▶ 偶联 探讨了偶联物的使用和控制,对于某些特定类型的合成寡核苷酸具有重要意义。 ▶ 溶液中的活性物质 针对溶液中的原料药提出了特殊要求,考虑了其在溶液状态下的稳定性和活性。 本指南全面覆盖了合成寡核苷酸从研发到生产、再到质量控制的各个环节,为制药行业提供了详细的指导原则,以确保合成寡核苷酸药物的安全、有效和质量可控。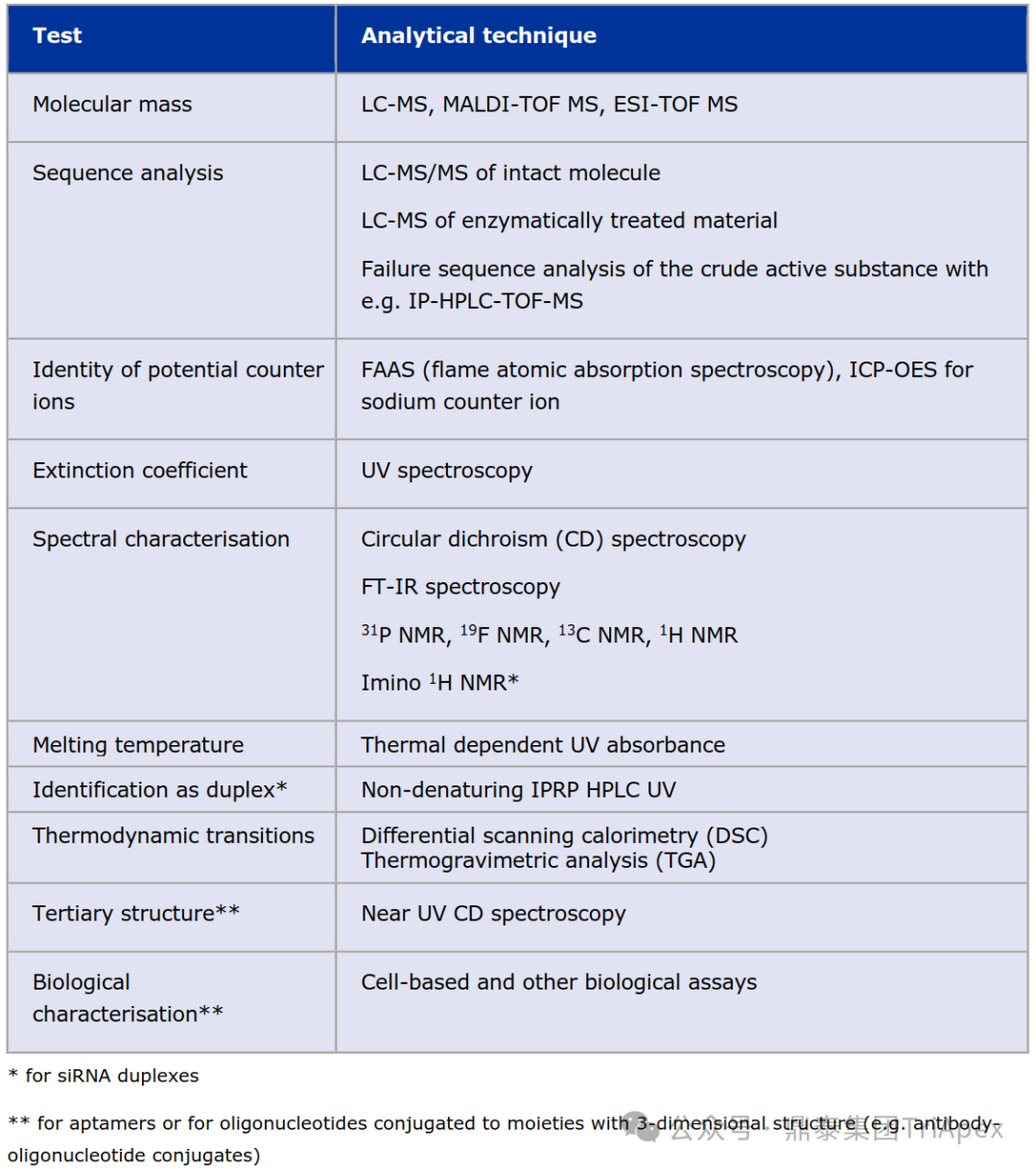
4. 对药物制剂的考量 详细阐述了含有寡核苷酸活性成分的药用制剂的质量目标产品概况(QTPP),这直接关联到药物的质量、安全性和有效性: ▶ 质量目标产品概况(QTPP) QTPP 考虑了药物的给药途径、剂型、生物利用度、强度和稳定性等因素,这些因素对于含有寡核苷酸作为活性成分的药物至关重要。 ▶ ICH 指南的应用 ICH Q3B 和 Q6A 指南不完全适用于寡核苷酸,但关于杂质的阈值讨论在 3.2.S.4.1 中提到的原则同样适用于最终的药物制剂。 药物制剂中形成的降解产物应设定限制,考虑到上述阈值和最大日剂量,这些限制需基于批次分析数据和确证数据进行个案评估。 ICH Q3D 适用于寡核苷酸,因此,对于含有寡核苷酸作为活性成分的药物产品,该指南中列出的要求是适用的。 对于用于人类使用的药物制剂中的寡核苷酸活性物质,关于亚硝胺杂质的风险考量和要求同样适用。 ▶ 潜在相互作用 应在生产期间评估寡核苷酸与处方中赋形剂以及来自起始物料和包装材料(如胶塞)可能产生的可浸出物之间的潜在相互作用。 ▶ 药物制剂的效力检测 如果药物的作用机制仅基于寡核苷酸的初级结构和含量(数量),则无需进行效力检测以进行药物制剂的放行和稳定性测试。 鼓励申请者提供更多关于可能存在的(或不存在的)高级结构的细节,例如基于近紫外 CD 光谱学或其他技术,以及在可行的情况下进行计算研究。 此外,还应进行关于高级结构稳定性的试验。
5. 仿制药开发(仅限人用产品) 对于合成寡核苷酸的仿制药开发,通常应遵循 <生物等效性指南 (CPMP/EWP/QWP/1401/98 Rev.1/Corr**)> 中描述的要求。对于复杂制剂(例如脂质纳米颗粒),可能需要额外的临床和/或非临床研究,此时应考虑在关于 < Data requirements for intravenous liposomal products developed with reference to an innovator liposomal product> 的思考文件中描述的基本原则。 ▶ 分析比较测试 使用广泛的一系列表征研究进行分析比较,这些研究适用于特定的寡核苷酸类别,构成了可比性研究的基础。 对于含有硫代磷酸酯连接的寡核苷酸,应进行额外的研究以确定其对映异构体的组成。由于硫代磷酸酯连接处的磷为手性中心,这些活性物质可能包含许多不同的对映异构体。应采用合适的最新分析方法进行立体化学表征研究。 申请人有责任证明纯度分析方法适用于覆盖寡核苷酸的完整杂质概况,或者是否需要通过额外的补充方法进行额外的纯度测试。当观察到与原研产品杂质概况存在差异时,应证明不在原研产品中存在的杂质已得到界定(qualified)且不引起任何安全担忧。杂质含量超过 1.0% 应进行鉴定,超过 1.5% 应进行界定。 ▶ 强制降解研究 推荐进行比较性强制降解研究,以证明纯度分析方法能够充分表征两种产品的杂质概况。 在可比性研究中使用的分析方法应是适用且经过充分验证的,并且灵敏度足以检测两种产品之间的潜在差异。如果使用统计模型来证明仿制药产品与原研药之间的可比性,应进行充分描述和证明。用于头对头对比分析的批次最好来自商业途径。用于可比性研究的批次数量应进行充分证明(参考:Reflection paper on statistical methodology for the comparative assessment of quality attributes in drug development - EMA/CHMP/138502/2017)。
6. 临床试验申请要求(仅限人用产品) 临床试验申请对于用于临床研究的寡核苷酸的要求会随着开发阶段的不同而变化,从早期到 Ⅲ期临床试验直至上市许可申请阶段,要求会逐渐增加。在早期阶段,主要关注点在于寡核苷酸的安全性。 ▶ 生产过程 固相合成:寡核苷酸的生产主要通过固相合成,因此可以借鉴类似寡核苷酸的开发经验。 具体细节:需要提供关于特定固相合成过程的信息,包括固相载体类型、活化剂、氧化/硫化剂及封端剂的使用情况。 冻干工艺参数:在后期临床开发阶段,需提供冻干过程的参数。 ▶ 起始物料 受保护的亚磷酰胺:作为活性物质的起始物料,在早期阶段就需要特别注意某些已知关键杂质。 质量标准说明:起始物料的质量标准应在 IMPD 中提供,但基于关键性评估设置某些杂质限制是在后期开发中才需要的。 ▶ 原料药后处理 脱保护与解离:对从固相载体上解离并脱保护后的粗寡核苷酸及其关键纯化步骤(如色谱法、超滤)同样有类似的期望。 双链寡核苷酸:对于双链寡核苷酸,应提供单链中间体的质量标准。 ▶ 生产工艺变更 变更描述:生产过程中的任何变更都应描述其对寡核苷酸质量的潜在影响。 关注关键杂质:特别是要注意与临床前批次相比,关键杂质水平的变化。 ▶ 产品特性 序列表征:寡核苷酸的序列应该完全表征;对于适配体等特殊结构,还需研究其高级结构。 聚集体:应调查聚合的可能性。 相关杂质:在开发过程中应对产品相关的杂质进行识别和/或鉴定。 分析方法:鼓励从早期阶段就开始采用正交分析程序,例如 AX-HPLC & RP-IP-HPLC 或结合正交原则的 HPLC-UV-MS。 ▶ 双链寡核苷酸 对于双链寡核苷酸,预期使用变性和非变性分析方法。 ▶ 适配体 生物活性测试:在 Ⅰ 期临床试验开始前,适配体的质量标准中应包含生物活性测试,基于适当、可靠和合格的方法。 ▶ 稳定性研究 分析方法:进行稳定性研究时,必需使用能够指示稳定性的分析方法。 储存条件:探索与寡核苷酸性质相符的储存条件。 参考经验:如果能被证明且有数据支持,可以参考其他具有相似化学性质和生产过程的寡核苷酸分子的稳定性信息来确定再检测期/保质期和储存条件。
7. N-of-1/个性化药物(仅限人用产品) 详细阐述了针对单一患者的个性化寡核苷酸治疗产品的特殊考虑: ▶ 适用范围 当寡核苷酸用于治疗全球唯一患有严重或威胁生命的罕见疾病的患者时,这类药物被视为个性化药物。 ▶ 鉴定 必须证明所采用的方法能够区分开发项目中使用的所有序列,以确保其序列的准确性。 ▶ 纯度与分析方法 对于原料药及其制剂,应开发适合的、先进的纯度分析方法,这些方法需具有稳定性指示特性。不建议仅使用一种方法(如离子交换色谱 [IEX] 或尺寸排阻色谱 [SEC] )来进行纯度检测。在开发过程中,应定期重新评估分析方法并根据新获得的知识进行必要的更新。 ▶ 杂质控制 检测总体纯度是强制性的,但对于仅用于单一患者的药物,产品相关杂质的研究可能较为有限。可以初步使用文献中发表的一般性杂质检测方法。 ▶ 稳定性研究 对于 N-of-1 治疗方案,不需要遵循 ICH Q1 稳定性指南进行全面稳定性研究。对于每个拟用于临床试验的寡核苷酸,需要提供稳定性数据。随着更多综合数据的积累,可以基于先前知识采取替代性方法。如果仅对活性物质进行稳定性测试,则需要证明这些数据对于药物制剂也是相关的,特别是考虑到预期的储存条件和提议的有效期。 ▶ 制剂选择 推荐选择与已批准产品类似的制剂。药物制剂的选择可能只需要有限的药剂学开发。 ▶ 微生物学控制 应开发微生物学控制策略,并确保经由注射途径给药的产品无菌。 ▶ 其他注意事项 基于公开信息而完全省略原料药和制剂的稳定性研究是不能接受的。关于 GMP 方面的问题,建议与负责生产地点的 GMP 监管机构进行讨论,包括材料重复使用(如树脂或超滤/透析滤器)和潜在交叉污染风险等问题。 由于作者非 CMC 专业,且化学合成寡核苷酸药物的开发和生产是一个较新的行业话题,加之时间有限,错误或不足之处在所难免。如有不准确或错误之处,敬请同行不吝指教。
扫码获取 EMA guidance 原文: 参考资料: [1] EMA. Concept Paper on the Establishment of a Guideline on the Development and Manufacture of Synthetic Peptides, 2022. [2] EMA. Guideline on the Development and Manufacture of Oligonucleotides (Draft), 2024.







 法规政策
法规政策 项目管理
项目管理


 Global
Global



 苏公网安备32011202001081
苏公网安备32011202001081