


上篇分享了RNA技术的最新进展、化学制造与控制两大议题(点击阅读原文),本篇文章将围绕 RNA 药物非临床研究中的关键性问题、行业和监管机构共同关注的“平台数据”和平台认定的科学依据、临床试验等方面的内容进行总结,并对 EMA RNA 药物指导原则的出台进行了展望。
★ 文章导览 ★
Session 1 | State of the Art of RNA Technologies |
Session 2 | Chemistry, Manufacturing, and Controls Aspects of RNA Technologies |
Session 3 | Nonclinical Aspects of RNA Technologies |
Session 4 | Clinical Aspects of RNA Technologies |
Session 5 | Panel Discussion |
关键词:非临床研究、挑战和机遇、相关种属/动物模型、NAM;平台数据、科学依据、决策树;临床试验、个性化治疗
Session 3: Nonclinical Aspects of RNA Technologies
(1) 非临床研究的挑战和机遇
RNA 药物非临床开发面临的主要挑战包括:如何识别和减轻在靶毒性和脱靶毒性,实现组织或细胞内的靶向递送。这对开发和应用合适的体外和体内模型提出了更高的挑战,尤其是当开发个性化RNA 药物时会显得尤为困难。迫切需要针对这一新型药物出台通过什么样的证据支持监管批准的指导原则。
Haiyan Zhou(UCL)报告了 RNA 药物非临床开发的机遇,如利用先进的基因组学技术发现新的治疗靶点,开发新的化学结构、作用机制和递送系统等。通常,可通过体外试验和动物体内试验来预测人体内的安全性和有效性。通过计算机预测(in silico)和体外试验(如 mRNA-seq)预测脱靶效应,通过 PBMC 孵育体系评估免疫反应,通过检测 RNA 转录、蛋白表达和功能检测评估体外药效学活性。由于很多目标序列是人体特异(human-specific)的,与动物靶点的同源性较低,因此常采用转基因动物评价体内药效学。因某些遗传性疾病具有人体特异性,新的非临床研究模型(NAMs),如来自人体的 2D 细胞模型和 3D 类器官,将变得非常重要。学术界与工业界之间的合作对于这些 NAMs 的开发至关重要。


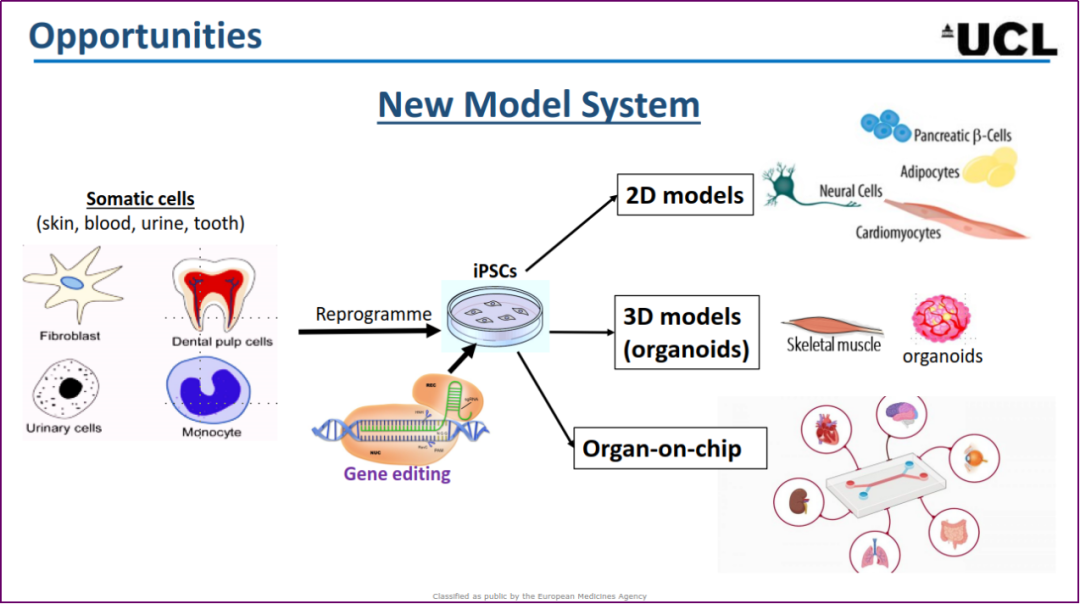
RNA 药物非临床研究的机会和挑战(引自Haiyan Zhou)
Camilla Svensson(MPA)从监管的角度解释了获取非临床研究数据(如了解药理活性、药代动力学和毒性信息)的重要性。与后文提到的Susan Goody(Moderna)的观点类似,在进行了科学论证的前提下,有时可以合理地利用已有的数据。Camilla 汇总了当前可参考的非临床指导原则,并阐述了 RNA 药物开发面临的机遇和挑战。机遇在于可遵循 3R 原则利用现有知识优化非临床研究计划;挑战包括对平台的定义、明确对具体产品的要求、理解新的递送载体/配体的异质性,以及药理学相关种属的可获得性。她还列出了非临床安全性评估中仍需解决的关键点:在靶毒性、脱靶毒性、化学结构依赖的类效应、制剂处方/偶联物的安全性、免疫原性/反应原性以及药代动力学。此外,鼓励申请人尽早与监管机构进行沟通交流。



RNA 药物非临床研究的机会和挑战(引自Camilla Svensson)
(2) 使用“平台数据”的科学依据
Susan Goody(Moderna)从工业界的角度介绍了简化 RNA 药物开发的可能性,并建议行业与监管机构进行合作,共同定义一些可充分利用非临床平台数据的关键要素,包括:药物结构特点(如RNA序列组成)、特定药物的非临床数据(如安全性、药代动力学和生物分布数据)以及利用平台数据的科学依据。
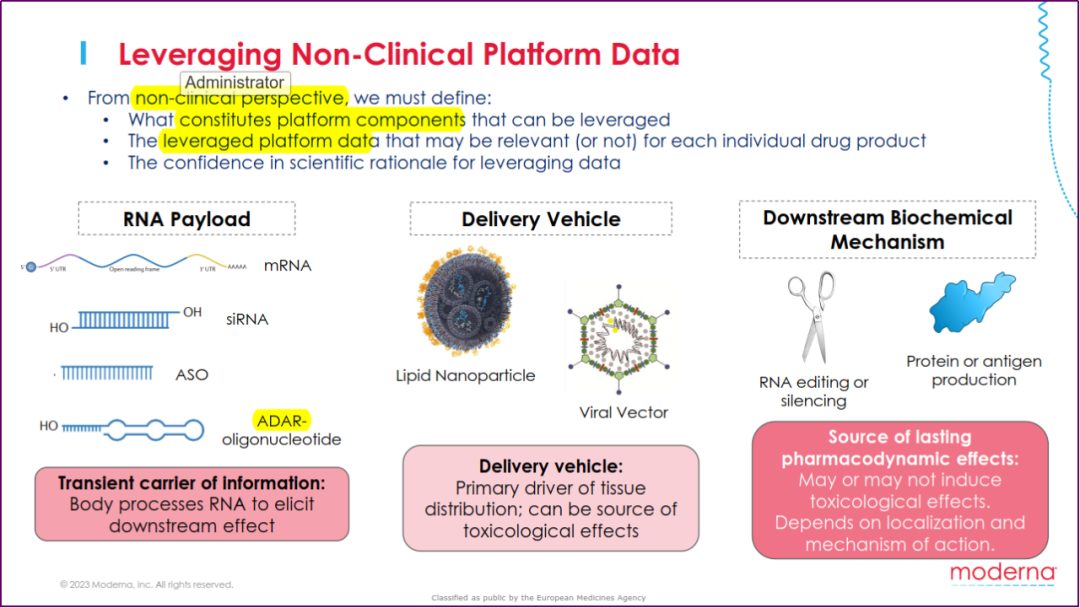
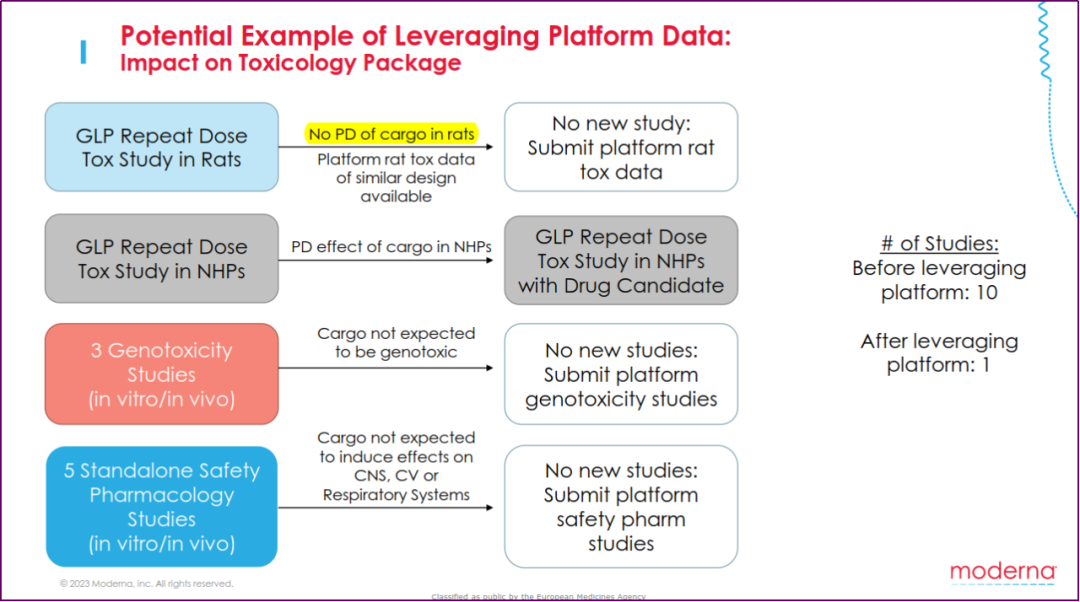
利用平台数据可简化非临床研究(引自Susan Goody)
Susan提出了一种可能的决策树,以帮助申请人和监管机构就如何利用非临床平台数据的科学依据达成一致,其中包括评估 RNA 下游药效作用是否可能影响非临床研究结果、是否使用了共同的平台元素以及是否有可靠的使用平台数据的科学依据。
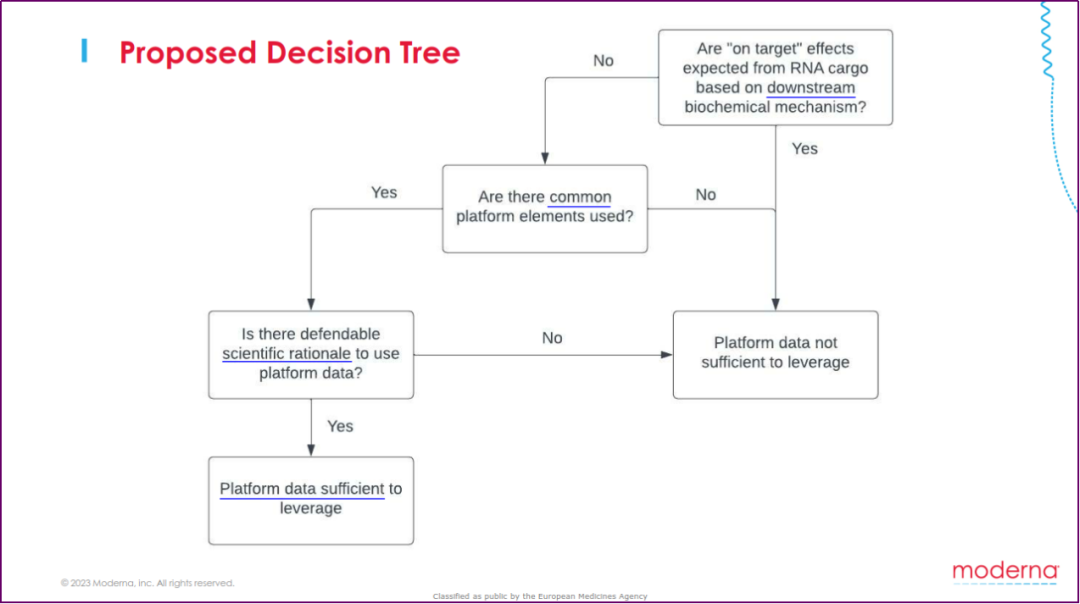
平台数据科学依据决策树(引自Susan Goody)
基于科学依据,采用平台数据有可能加速药物开发进程和治疗手段的可及性。通过对比采用和不采用平台研究的非临床毒理学/安全药理学的案例发现,采用平台数据有助于缩短非临床研究的时间、优化动物使用和提高申报资料准备效率。
应根据对特定化合物的已知程度,基于Case by case原则对平台数据的适用性进行评估。如果平台质量属性(如纳米颗粒的粒径)发生了变化,是否需要额外的非临床分析取决于对相关信息的了解程度,以及是否有基于科学数据的信心认为这种变化不会对新产品的评估造成影响。
(3) 相关种属/动物模型
动物模型对于评估药物安全性至关重要。可通过平台方法和/或源自人体的细胞模型减少动物的使用并支持临床试验。NAM 应经过验证并证明能用于评估供试品的安全性特征。建议对 NAM 进行资格验证以得到监管机构的认可。
每种模型均有其局限性,需要根据疾病/药物进行调整和优化。某些情况下,如基于 RNA 的治疗性肿瘤疫苗,可能没有适当的动物模型用于评估其安全性。EMA 也意识到,某些特定产品的开发缺乏相关动物模型。
Session 4: Clinical Aspects of RNA Technologies
该会议讨论了 RNA 疗法临床试验的过去、现在和未来面临的挑战,如临床前研究向临床试验的转化、剂量确定、临床与实验室之间的去而复返和迭代(iterating between clinic and laboratory)、毒性和长期安全性。
David Henshall (Royal College of Surgeons)从学术角度阐述了癫痫领域的研究进展,并分享了 RNA 疗法应用于脑部疾病的案例。既往的癫痫治疗方法并未针对基础病理生理机制,需开发针对单基因病因学和继发性(如脑损伤后)癫痫的新疗法。RNA 药物可能适用于上述两类癫痫患者的功能丧失或突变增加。
很多患者寻求基于 RNA 的个性化治疗,有助于避免频繁给药和严重的副作用。同时,患者也对这些药物疗效的持久性以及特殊给药途径(如脊髓注射)的安全性有所担心。从转化医学的角度来看,RNA 药物面临的挑战包括:不易通过血脑屏障,不清楚哪种化学性质能获得最佳的耐受性和疗效,以及如何预测 RNA 药物的脱靶效应和与标准治疗药物的相互作用。同样,从转化角度考虑,可能的机遇是利用手术后癫痫患者的脑组织来测试RNA 药物;循环中 RNA 水平可用作患者富集的生物标志物(即伴随诊断)和用于考察 PK 特征。某些种系的犬患有抗药性癫痫,可尝试采用犬建立模型用于在人临床试验之前考察疾病进程或用于指导兽药的开发。
Michael Wenger(BioNTech)分享了 RNA 药物在肿瘤学和免疫学领域的研究经验,并从工业界角度分析了临床研究面临的挑战。针对癌症的 RNA 免疫疗法主要包括使用 mRNA 在癌症患者体内表达“现成”的固定抗原或针对肿瘤新生抗原(neoantigens)的个性化抗原。选择新生抗原需要基于不断进化的“学习型”算法,这将面临着静态监管方面的挑战。并提出了 mRNA 技术用于在患者体内表达抗体和细胞因子(即“蛋白替代”)的展望。
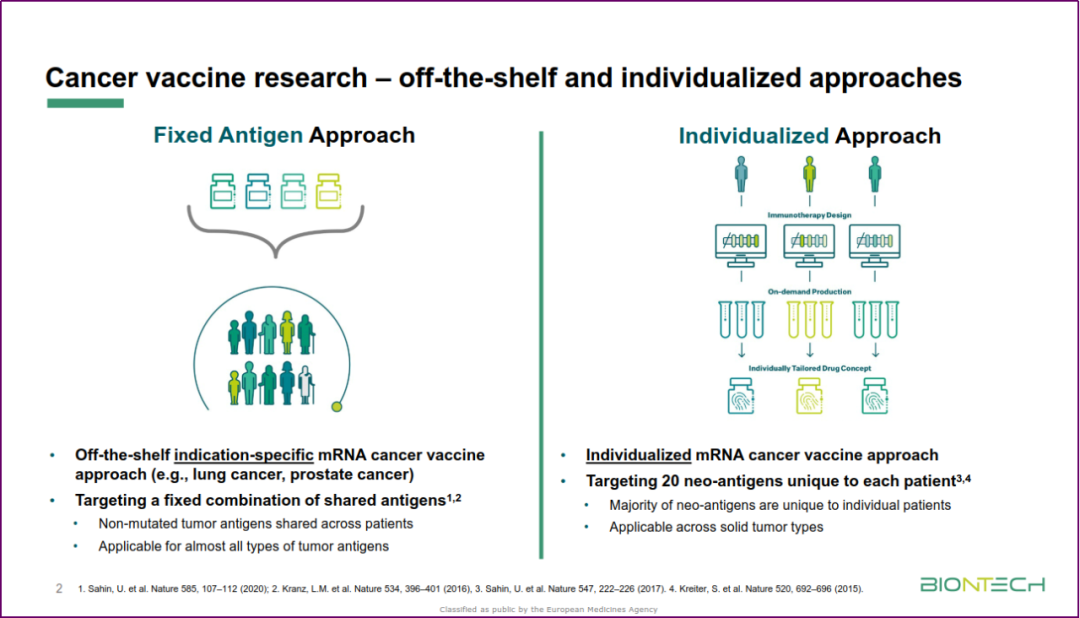
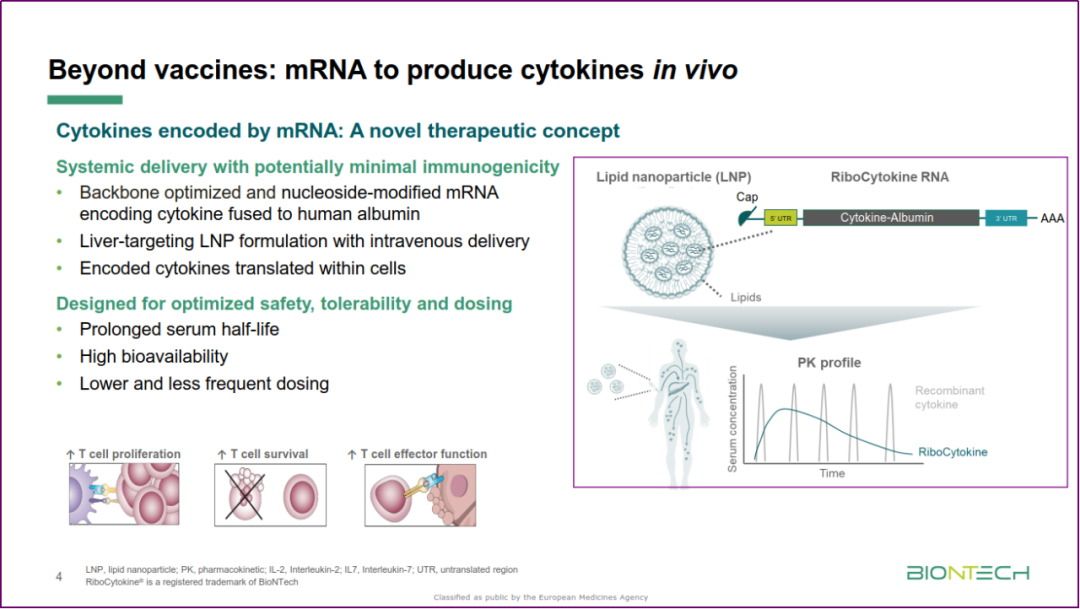
mRNA 疗法的新应用(引自Michael Wenger)
Joop van Gerven(CCMO)详细介绍了伦理和国家主管机构对 RNA 疗法临床研究的看法。CCMO评估了超过100项 RNA 疗法的临床试验申请,其中以 ASOs 和 siRNAs 最为常见,也包括剪接修饰剂、编码 mRNA 等,约 8% 因获益/风险证据不足而未被批准。RNA药物的加速批准得益于良好的药理学依据、药代动力学和组织渗透性,以及早期开发中确立了可用于剂量选择的生物标志物。早期涉及儿科人群的试验,如果仅关注药代动力学和安全性可能是不够的;而如果评估了临床活性/疗效(的生物标志物),则审批可能会更顺利。
讨论环节涉及加快 RNA 药物研发的合作、资金、平台试验、患者参与、合作伙伴关系、物流和相关统计学方法等。
Session 5: Panel Discussion
Arjon Van Hengel (DG Research and Innovation EC)强调了数据共享和针对未满足临床需求开展研究的重要性。
Mariette Driessen (VSOP)强调了对患者和公众进行教育的重要性,尤其是将新的 RNA 技术应用于罕见病(特别是儿科患者)时。
Jeske Smink(Silence)表达了对更多监管指导和不同成员国和地区之间协调一致的期待,如关于杂质分类的指南。建议制定更多灵活的指导原则,以适应 RNA 药物的持续发展或适应各种 RNA 模式的开发。促进不同利益相关方对话,以支持 RNA 药物的开发。
Q
A
问:How can consortia and patient organisations get in contact with regulators and what can regulators do for them?
答:患者组织和合作组织可以通过参与平台活动和组织网络会议与监管机构建立联系。在荷兰,有一个专门的平台可以让患者组织能够分享信息并接触政策制定者,特别是在基因治疗领域。患者在与开发者的对话中扮演着关键角色。学术界面临的一个挑战是缺乏监管事务的专业知识。对于起草个体化治疗(如基于突变的产品)的监管文件尚缺乏经验,这些挑战只能通过患者的参与来克服,患者的参与可以使临床试验更好地满足其需求。
问:Could Q&A documents be a quick solution to the lack of guidance/guidelines (which would require more time to be completed)?
答:Q&A文件在早期发展中非常有用,可以为 RNA 治疗方式提供具体且恰当的指导。利益相关方可以文献知识或经验为依据,通过EMA 的 Innovation Task Force (ITF) 或Scientific Advice (SA)提出提案和挑战监管机构。监管机构有意愿在认为有价值的时候支持并起草指导原则。
问:For platform approaches, when are products considered similar?
答:为了建立有意义的平台方法,需要在申请人和监管方面同时设立标准。
申请人方面,需提供证据证明平台方法确保了同类产品的安全性和有效性;需要对RNA分子在不同病症和不同作用方式下使用积累更多的经验和更深度的理解;在申报资料中,通过平台方法获得的部分数据可用于特定的环节,如质量。有些平台是公司专属(company-specific)的,而有的则是相似的或具有相同的作用机制。需要有相应的标准用于定义平台和类似产品(similar products)。
问:What is the main point necessary to move forward with RNA-based medicines?
答:强调了共同核心数据对于推进 RNA 药物临床试验的重要性,以及早期不同利益相关方之间的早期、保持适度的监管灵活性的必要性。
结语
RNA 药物在许多目前缺乏治疗方法的疾病中具有巨大潜力,也提出了多个科学和监管方面的挑战。当前,学术界在注册申报方面的专业知识不足,监管机构在 RNA 药物方面的指南文件存在空白,特别是在质量方面;平台方法的确立和接受标准仍存在挑战。通过本次会议收集的知识有助于为罕见病、神经系统疾病和免疫系统疾病以及癌症等关键领域新技术药物指导原则的制定提供依据,EMA表示将考虑会议中提出的建设性意见,以推进未来指导方针和问答文档的制定。
EMA RNA 药物指导原则,还远吗?我们满怀期待。
参考资料:
[1] Falk Ehmann, Andreas Kuhn, Anna Maria Gerdina Pasmooij, et al.
Report of the European Medicines Agency Conference on RNA-Based Medicines[J]. Nucleic Acid Ther . 2024 Feb;34(1):4-11.
doi: 10.1089/nat.2023.0021. Epub 2024 Jan 4.
扫码添加好友
获取 EMA《 Regulatory and scientific virtual conference on RNA-based medicines》会议资料







 法规政策
法规政策 项目管理
项目管理


 Global
Global




 苏公网安备32011202001081
苏公网安备32011202001081